
ABSTRACT
IRGM is an established genetic risk factor for Crohn disease (CD) and several other inflammatory disorders. However, the mechanisms employed by IRGM to restrain the inflammation are not known. In our recent study, we showed that IRGM negatively regulates NLRP3 inflammasome activation. IRGM employs 2 parallel approaches to constrain inflammasome activation. First, IRGM directly interacts with NLRP3 and PYCARD/ASC, and mediates their SQSTM1/p62-dependent macroautophagic/autophagic degradation. Second, IRGM impedes inflammasome assembly by blocking the polymerization of NLRP3 and PYCARD. We also found that IRGM suppresses NLRP3-mediated exacerbated outcomes of dextran sodium sulfate (DSS)-induced colitis in a mouse model. Taken together, this study presents evidence that IRGM can directly regulate inflammation and protect from inflammatory diseases.
The genome-wide association studies (GWAS) conducted by Wellcome Trust Case Control Consortium identified single nucleotide polymorphism’s (SNP’s) in the IRGM (Immunity-related GTPase M) locus to be strongly associated with Crohn Disease (CD). In another study by McCarroll et al. (2008), showed that a 20-kb deletion polymorphism in the IRGM promoter that reduces its expression is important for its genetic linkage with CD. Later, several studies performed by different groups, in different populations, from all over the world found association of CD or inflammatory bowel disease (IBD) with SNP’s in the IRGM locus. IRGM deficiency is also genetically and functionally linked with other inflammatory and autoimmune diseases including autoimmune thyroid diseases, Graves disease, ankylosing spondylitis, Sjogren syndrome, experimental autoimmune encephalomyelitis, hepatic steatosis and non-alcoholic fatty liver disease. NOD2 (nucleotide binding oligomerization domain containing 2) and ATG16L1 (autophagy related 16 like 1) are other well-established risk factors for CD. In our previous study, we showed that all 3 (IRGM, ATG16L1, and NOD2) CD genetic risk-factors physically interact with each other, where IRGM acts as a scaffold for the interaction of the microbial sensor NOD2 and autophagy proteins including ATG16L1, BECN1, and ULK1 (unc-51 like autophagy activating kinase 1) for antimicrobial defense. This study for the first time described the mechanism utilized by IRGM to regulate xenophagy; however, whether IRGM has any direct role in the regulation of inflammation was not clear. In our recent study [Citation1], we found that IRGM is a negative regulator of IL1B/IL-1β production by inhibiting NLRP3 inflammasome activation (). This study establishes a direct role of IRGM in inflammation regulation.
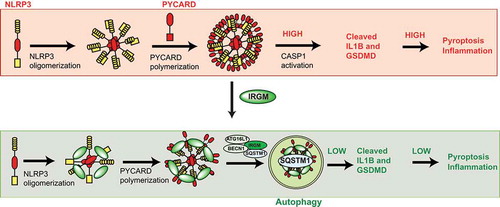
Figure 1. Graphical representation of the work. In the absence of IRGM, an increased number of NLRP3 inflammasomes are formed leading to enhanced activation of CASP1, IL1B and GSDMD resulting in augmented pyroptosis and inflammation. IRGM, when present, interacts with NLRP3 and PYCARD and inhibits inflammasome formation and also mediate SQSTM1/p62-dependent selective autophagy of inflammasomes, resulting in reduced levels of CASP1, IL1B, GSDMD, pyroptosis, and inflammation.
The NLRP3 inflammasome is one of the best-studied inflammasomes. Upon activation, NLRP3 and its adaptor molecule, PYCARD (PYD and CARD domain containing) oligomerize to form inflammasomes that can elicit conversion of proCASP1 into active cleaved CASP1. The activated CASP1 now can proteolytically cleave precursors of IL1B, IL18 and GSDMD (gasdermin D). GSDMD forms pores in the plasma membrane through which IL1B and IL18 are secreted out of the cells. This innate immune response to microbial stimuli is important to clear the invading pathogen. However, the aberrant activation of the NLRP3 inflammasome and chronic cytokine response is associated with the pathogenesis of CD and several other inflammatory diseases including gout, type 2 diabetes, cancer, cardiovascular diseases, Alzheimer, Parkinson, and prion diseases. It is now well understood that therapeutic targeting of the NLRP3 inflammasome can provide novel treatments for inflammatory diseases. Hence, it is important to understand the host mechanisms that can restrain the activation of the NLRP3 inflammasome and IL1B production.
In our recent study, we found that IRGM, whose expression is induced by pathogen-associated molecular patterns and microbes, is a negative regulator of transcription of pro-inflammatory cytokines (IL1B, IL18, and TNF/TNF-α). IRGM suppresses the NFKB/NF-κB and MAPK/p38 signaling pathways to control the expression of these cytokines. Currently, the mechanism used by IRGM to suppress NFKB and MAPK/p38 signaling pathways are not known. We also found that IRGM suppresses not only the production of pro-IL1B but also its cleavage. We found that IRGM restrains IL1B maturation by downregulating the NLRP3 inflammasome activity. Mechanistically, we first found that IRGM directly complexes with NLRP3 and PYCARD and, by binding to their oligomerization domains, it obstructs the polymerization of NLRP3 and PYCARD, leading to compromised formation of productive inflammasomes (). Second, IRGM mediates SQSTM1/p62-dependent selective autophagy of NLRP3 and PYCARD leading to reduced inflammasome numbers in the cell (). We performed several experiments to understand whether these 2 approaches used by IRGM are dependent or independent of each other. We found that inhibition of autophagy restores the IRGM-mediated degradation of NLRP3-PYCARD but is not able to rescue the oligomerization defect of NLRP3-PYCARD. Further, a GTPase activity mutant (S47N) of IRGM, which is defective in invoking autophagy, is not able to degrade NLRP3 but is able to reduce the oligomerization of NLRP3. These and several other pieces of evidence presented in the study suggest that IRGM utilizes 2 parallel independent approaches to control the activity of NLRP3 inflammasomes. Uncontrolled activation of the inflammasome can lead to pyroptosis, an inflammatory cell death. We observed increased pyroptosis in IRGM knockdown cells marked by increased propidium iodide staining of the cells and increased GSDMD cleavage. Taken together, our study revealed one of the important IRGM-mediated anti-inflammatory immune homeostasis mechanism by which IRGM can be protective against inflammatory diseases.
In the past few years, autophagy has emerged as one of the prominent mechanisms to control inflammation. Autophagy-mediated degradation of pattern recognition receptors and inflammatory signaling pathway intermediates averts the chronic activation of inflammation. NLRP3, DDX58/RIG-I, TMEM173/STING, CGAS, and AIM2 are some examples of inflammatory proteins that are degraded by autophagy to keep IL1B and type 1 interferon cytokines under check. The IRGM associates with SQSTM1/p62 and mediates ATG5- and ATG7-dependent canonical autophagy of NLRP3 and PYCARD (). Given the protective role of IRGM in interferonopathies, it will be interesting to determine whether IRGM can mediate degradation of pattern recognition receptors involved in interferon production.
Given the association of both IRGM and NLRP3 with IBD and CD, we employed the DSS-induced colitis mice model to understand physiological significance of above mentioned regulation. Like its human counterpart, mice IRGM1 also negatively regulates expression and oligomerization of NLRP3 and PYCARD leading to reduced inflammasome activation. We observed that DSS-treated irgm1 knockout mice develop intensified inflammatory symptoms such as a decrease in body weight, increased diarrhea, blood in stool and shortening of colon length compared to wild type. Inhibition of NLRP3 using a specific inhibitor, MCC950, mitigates all the increased inflammatory symptoms in irgm1 knockout mice suggesting that NLRP3 inflammasome activation in irgm1 knockout mice plays a significant role in the exacerbated outcome of the disease. Hence, administration of NLRP3 inhibitors in IRGM-deficient IBD patients could be a useful approach to reduce the severity of disease and symptoms.
Acknowledgments
This work is supported by the Wellcome Trust/Department of Biotechnology (DBT) India Alliance (IA/I/15/2/502071) fellowship, ILS core funding (Department of Biotechnology, India), and Early Career Reward (SERB, ECR/2016/000478) to Santosh Chauhan. Subhash Mehto is supported by a fellowship from SERB (NPDF, PDF/2016/001697). Swati Chauhan is supported by a fellowship from DST (SR/WOS-A/LS-9/2016).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Reference
- Mehto S, Jena KK, Nath P, et al. The Crohn’s disease risk factor IRGM limits NLRP3 inflammasome activation by impeding its assembly and by mediating its selective autophagy. Mol Cell. 2019 Feb 7;73(3):429–445.e7.