
ABSTRACT
Many cancer therapies operate by inducing double-strand breaks (DSBs) in cancer cells, however treatment-resistant cells rapidly initiate mechanisms to repair damage enabling survival. While the DNA repair mechanisms responsible for cancer cell survival following DNA damaging treatments are becoming better understood, less is known about the role of the epigenome in this process. Using prostate cancer cell lines with differing sensitivities to radiation treatment, we analysed the DNA methylation profiles prior to and following a single dose of radiotherapy (RT) using the Illumina Infinium HumanMethylation450 BeadChip platform. DSB formation and repair, in the absence and presence of the DNA hypomethylating agent, 5-azacytidine (5-AzaC), were also investigated using γH2A.X immunofluorescence staining. Here we demonstrate that DNA methylation is generally stable following a single dose of RT; however, a small number of CpG sites are stably altered up to 14 d following exposure. While the radioresistant and radiosensitive cells displayed distinct basal DNA methylation profiles, their susceptibility to DNA damage appeared similar demonstrating that basal DNA methylation has a limited influence on DSB induction at the regions examined. Recovery from DSB induction was also similar between these cells. Treatment with 5-AzaC did not sensitize resistant cells to DNA damage, but rather delayed recruitment of phosphorylated BRCA1 (S1423) and repair of DSBs. These results highlight that stable epigenetic changes are possible following a single dose of RT and may have significant clinical implications for cancer treatment involving recurrent or fractionated dosing regimens.
Introduction
Numerous cancer therapies rely on inducing double-strand breaks (DSBs) in cancer cells to induce cell death. However, in cases of treatment resistance, a subpopulation of cells is capable of repairing genomic lesions to survive treatment either via non-homologous end joining or homologous recombination. While a clear picture for the role of DNA repair proteins in this process has emerged (reviewed in [Citation1]), there is less clarity regarding the epigenetic mechanisms involved. In particular, the role of the basal epigenetic environment in influencing susceptibility to DNA damage, as well as changes to the epigenetic landscape following DNA damage, are not well characterized. The epigenetic changes in response to DNA damage are also key to understanding whether DNA methylation changes that result from radiotherapy (RT) may contribute to treatment resistance.
The chromatin landscape within tumour cells has the potential to influence whether they are susceptible to DNA damage caused by RT (reviewed in [Citation2]). It is conceivable that DNA in a loosely packed chromatin environment might be more susceptible to DNA damage than in a tightly condensed heterochromatin state. Conversely, damaged DNA in euchromatin could be expected to be more accessible to DNA repair proteins than when present in heterochromatin [Citation3]. Following signalling to recruit DNA repair proteins, the region surrounding the lesion must be remodelled to enable access. Following DNA repair, the local chromatin environment should ideally be restored to its original structure and epigenetic state. Therefore, an adaptive epigenetic response to DNA damaging treatments may have important implications for future response to treatment. In this context, evidence is emerging for heritable epigenetic silencing in regions surrounding DSBs [Citation4–Citation6] that could in turn influence susceptibility to DNA damage and treatment success. This is particularly important in the case of current cancer treatment regimens as patients receive repeated fractionated doses of RT and/or recurrent infusions of chemotherapeutic agents.
The most frequently characterized epigenetic alteration triggered by DNA damage is the formation of γH2A.X foci. These foci form when the histone variant H2A.X is phosphorylated at serine 139 in response to DSBs [Citation7] and appears important for recruitment of the relevant repair proteins. γH2A.X marks chromatin domains impacted by DNA damage and helps to initiate checkpoint-mediated arrest, spreading over large domains to mark the region for repair. However, the remodelling events and modifications to the epigenome prior to and following DSB repair are yet to be elucidated. DNA methylation is the most widely studied epigenetic modification and has been associated with RT treatment resistance, with demethylating agents improving therapeutic sensitivity of tumour cells [Citation8–Citation12]. Importantly, basal methylation status may provide clinically relevant predictors for response to treatment. DNA methylation may limit RT-induced DNA damage due to heterochromatin forming a protective environment. The mechanism by which DNA methylation influences susceptibility to DNA damage and subsequent repair remains to be determined.
Using an in vitro model of radiation response with LNCaP (radiosensitive) and PC-3 (radioresistant) prostate cancer cell lines, we have previously established a role for opposing regulation of DNA repair pathways, and in particular homologous recombination, at the transcriptional level in prostate cancer cells with opposing response to RT [Citation13]. A question that remains is whether cells utilized in this model exhibit an epigenetic response to this treatment. In this study, DNA damage, repair and DNA methylation changes were profiled prior to and following induction of DSBs in prostate cancer cell lines with varying sensitivities to DNA damage. Our analysis demonstrates that DNA methylation remains largely unchanged following a single dose of RT with the exception of a very small number of sites. We also reveal that treatment with a DNA hypomethylating agent delays recruitment of the active BRCA1 DNA repair enzyme and recovery from DNA damage.
Results
Cells with divergent response to radiotherapy display distinct basal DNA methylation profiles
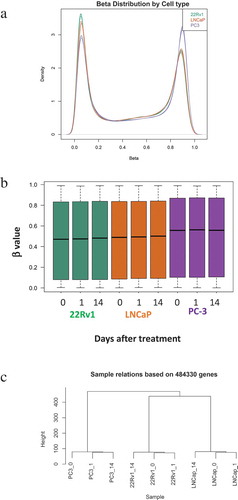
To evaluate how RT may influence the epigenome, DNA methylation profiles of prostate cancer cells were determined using the Illumina Infinium HumanMethylation450 BeadChip platform (Illumina HM450K arrays). DNA was extracted from untreated cells at 1 or 14 d following a single radiation dose (2 Gray (Gy)) to determine both short-term response and more stable changes. Included in this analysis were LNCaP, 22Rv1 and PC-3 cells, derived from a lymph node metastasis, primary prostate tumour and bone metastatic disease, respectively. We have shown that these cell lines vary in terms of radioresponse with the LNCaP cells being radiosensitive, the 22Rv1 cells displaying intermediate radioresponse and the PC-3 cells being radioresistant ([Citation13], Supplementary Figure 1) as demonstrated using clonogenic assays. At these doses of radiation induction of apoptosis was observed, however there was no significant difference between cell lines (Supplementary Figure 2). Beta (β) values were used to measure levels of DNA methylation, these range from 0 to 1, with 0 representing unmethylated CpGs and 1 representing fully methylated CpGs. Analyses indicated distinct DNA methylation patterns between the three cell lines. Overall, PC-3 cells had a larger proportion of hypermethylated probes as determined by β values, compared to the LNCaP and 22Rv1 cells (). Hierarchical clustering based on methylated probes resulted in each cell line clustering distinctly from each other () with the methylation profiles obtained for the more radiosensitive 22Rv1 and LNCaP cells being more closely related than the radioresistant PC-3 methylome.
Figure 1. Methylation profiles of LNCaP, 22Rv1 and PC-3 cell lines before and after radiotherapy. Prostate cancer cell lines were exposed to 2 Gy radiation and DNA was extracted at 0, 1 and 14 d. DNA methylation was profiled using the Illumina Infinium HM450K platform. (a) Density distribution of β values for the LNCaP, 22Rv1 and PC-3 cell lines. (b) β value distribution for the three cell lines and time-points. (c) Sample relatedness ranked according to methylation status across the cell lines and time-points.
DNA methylation stability in prostate cancer cells following radiotherapy
Following from the analysis of basal DNA methylation profiles in the three cell lines, we next sought to determine how DNA methylation may be altered by exposure to RT. DNA methylation profiles of untreated cells versus those exposed to RT were compared. Overall, DNA methylation was found to be largely unchanged by RT treatment. However, a number of CpG sites had a > 0.25 change in β value over the time course of treatment () with 39 altered CpG sites detected in the LNCaP cells, 33 in the 22Rv1 cells and 35 in the PC-3 cell line. This represents approximately 0.01% of the HM450K array coverage, and these CpG sites were random in terms of distribution being detected across numerous chromosomes and located in gene bodies, promoters, transcription start sites and 5ʹ UTRs. While a small portion of these CpG sites became hypomethylated by day 14 post RT, the vast majority displayed increased DNA methylation (). Two CpG sites were altered in response to RT in more than one cell line; ZMIZ1 (cg14371731) in LNCaP and PC-3 cells and HOXA11 (cg27309564) in the LNCaP and 22Rv1 cell lines. Interestingly methylation at these sites did not follow the same pattern of change for each cell line over the time course.
Figure 2. Methylation changes following radiotherapy in LNCaP, 22Rv1 and PC-3 cells. Prostate cancer cell lines were exposed to 2 Gy radiation and DNA was extracted at 0, 1 and 14 d. DNA methylation was profiled using the Illumina Infinium HM450K platform. (a) Heatmaps (generated using Morpheus https://software.broadinstitute.org/morpheus/) based on β values of differentially methylated regions post-radiotherapy in LNCaP, 22Rv1 and PC-3 prostate cancer cells at 0, 1 and 14 d. β values range from 0 to 1 with 0 being unmethylated (blue) and 1 being 100% methylated (red). (b) Bisulphite sequencing validation of methylation for the HOXA11 gene body for six clones (Illumina probe cg27309564 spans last three CpGs highlighted in box) in 22Rv1 cells. White circles represent unmethylated CpGs and black circles represent methylated CpGs.

HOXA11 methylation has previously been identified as a potential predictive marker for poor outcomes in acute myeloid leukaemia (AML) and ovarian cancer [Citation14,Citation15] and was found to have altered DNA methylation following a single dose of RT in both LNCaP and 22Rv1 cells. 22Rv1 cells were chosen for validation as this cell line is derived from the primary tumour, which is the site receiving RT and is therefore most clinically relevant. In the 22Rv1 cells, the β values for 0, 1 and 14 d for the HOXA11 cg27309564 probe were 0.228, 0.487 and 0.609, respectively, indicating an increase in methylation over the time course. To validate this data, DNA from non-irradiated and irradiated 22Rv1 cells was subject to bisulphite conversion and examined by clonal sequencing across the HOXA11 gene body (). The increased methylation evident at this HOXA11 site following RT uncovered by the arrays was reflected in the methylation profile observed by clonal bisulphite sequencing.
No differences in susceptibility to DNA damage were detected between sensitive and resistant prostate cancer cells
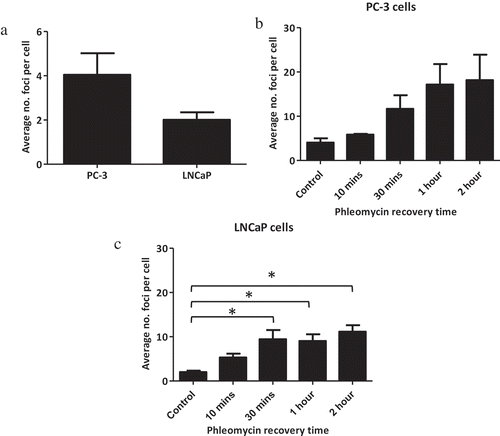
While the Illumina HM450K array data indicated that each cell line had a distinct DNA methylation profile, it was unclear as to whether this may reflect inherent differences in their susceptibility to DNA damage. To determine whether there were innate differences in sensitivity to DNA damage between cell lines, the cell lines with the two extremes of RT response, LNCaP (radiosensitive) and PC-3 (radioresistant) cells were treated with the radiomimetic phleomycin to induce DSBs. This compound generates DSBs similar to those induced by ionizing radiation via a free-radical driven process [Citation16]. Following treatment, cells were allowed to recover for 10 min, 30 min, 1 h or 2 h before undergoing γH2A.X immunofluorescence staining. PC-3 cells displayed an average of 4 γH2A.X foci per cell with reduced numbers of foci at baseline in LNCaP cells, averaging 2 foci per cell (), although this difference was not statistically significant (P = 0.119). Next, we profiled the extent of DNA damage and recovery in these cells, by determining the resolution of γH2A.X foci following DNA damage. PC-3 () and LNCaP cells () displayed a similar trend with γH2A.X foci increasing during the 2-h recovery period. While these changes were not significant for the PC-3 cells (P = 0.063), LNCaP cells displayed a significant increase in foci over the time course (P = 0.008). Interestingly the radioresistant PC-3 cells consistently displayed a higher number of foci than the LNCaP cells. Untreated PC-3 cells averaged 4 foci per cell, increasing to an average of 18 foci per cell 2 h following DNA damage (). LNCaP cells averaged 2 foci per cell when untreated and by 2 h following exposure to phleomycin, this had increased to an average of 11 foci per cell. However, the differences in foci numbers between the cell lines were not significant. Using the prolonged presence of γH2A.X foci as an indirect marker for damage recovery, these data demonstrated that the LNCaP and PC-3 cells responded to damage in a similar manner within a 2-h time frame. In addition, the PC-3 cells (radioresistant cell line) appear to experience slightly higher levels of initial DNA damage compared to the LNCaP cells (radiosensitive) under the same treatment.
Figure 3. Basal susceptibility to DNA damage and DNA damage recovery in sensitive and resistant prostate cancer cell lines. PC-3 and LNCaP cells were grown on chamber slides and exposed to phleomycin for 1 h and left to recover for the indicated times. Cells were then probed for γH2A.X foci via immunofluorescence staining and the average number of foci for 100 cells determined. (a) Basal levels of DNA damage in LNCaP cells and PC-3 cells. (b) Foci counts for PC-3 cells following phleomycin recovery. (c) Foci counts for LNCaP cells following phleomycin recovery. The experiments were performed in triplicate and the average number of foci for 100 cells determined. The mean and SEM for three biological replicates are shown. Statistical significance was determined using Student’s t Test (a) or One-way ANOVA (b & c) *P < 0.05.
Treatment with 5-azacytidine delays DNA repair in PC-3 cells
While the prostate cancer cell lines displayed distinct basal DNA methylation profiles, there were limited differences in terms of changes to DNA methylation following RT and their inherent susceptibility to DNA damage. The role of DNA methylation in the DNA damage response was therefore further investigated by attempting to reduce the levels of DNA methylation in the PC-3 cells, which are more resistant to DNA damage [Citation13] and displayed higher levels of basal DNA methylation (). A number of previous studies have demonstrated the sensitizing capabilities of the demethylating agent 5-AzaC which has been shown to increase cell death [Citation9,Citation10,Citation17]. In order to assess whether decreasing global DNA methylation could augment DNA damage and cell death in response to the radiomimetic, PC-3 cells were treated with 5-AzaC prior to DSB induction. Survival was profiled via clonogenic assay comparing untreated cells to those pre-treated with 5-AzaC before exposure to phleomycin. No significant difference in colony formation was observed between cells treated with phleomycin alone and cells pre-conditioned with 5-AzaC before phleomycin treatment (). This suggests that 5-AzaC did not alter the survival of prostate cancer cells following DNA damage. Reduced DNA methylation in the 5-AzaC-treated cells was confirmed by methylation-sensitive restriction digestion (data not shown). Next, we measured the number of γH2A.X foci in cells treated with or without the DNA hypomethylating agent in combination with phleomycin. While PC-3 cells treated with 5-AzaC exhibited a lower number of γH2A.X foci at 2 h, by 24 h the number of foci was significantly higher in comparison to the control cells (P = 0.0143, and γH2A.X panels in ).
Figure 4. Influence of 5-AzaC on cell survival and DNA repair. PC-3 cells were either pre-treated with 5-AzaC or left untreated, then treated with phleomycin for 1 h and left to recover. (a) Clonogenic assay showing cell survival with or without 5-AzaC pre-treatment. The mean and SEM for three biological replicates are shown. (b) Average foci counts for γH2A.X staining. The mean and SEM for three biological replicates are shown. Statistical significance was determined using Two-way ANOVA, *P < 0.01. (c) Representative example for immunofluorescence staining of DNMT1 and γH2A.X in cells treated with or without 5-AzaC prior to phleomycin addition.
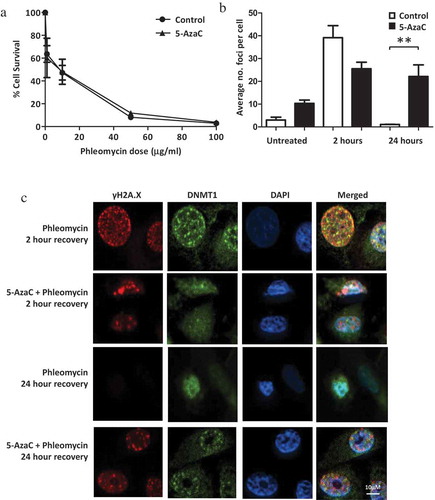
DNA methylation is maintained by DNMT1 which has previously been demonstrated as being recruited to DSBs [Citation18], therefore we were interested in determining how recruitment of this enzyme was influenced by treatment of cells with 5-AzaC prior to DSB induction. Using immunofluorescence staining it was found that nuclear DNMT1 levels appear higher while DSBs are present. While there is some co-localization evident, the enzyme does not appear to completely co-localize with the DSBs (). More punctate DNMT1 staining is seen in cells with DNA damage as opposed to those which have undergone repair (24-h post-treatment with phleomycin).
5-azacytidine treatment delays BRCA1 recruitment to DSBs
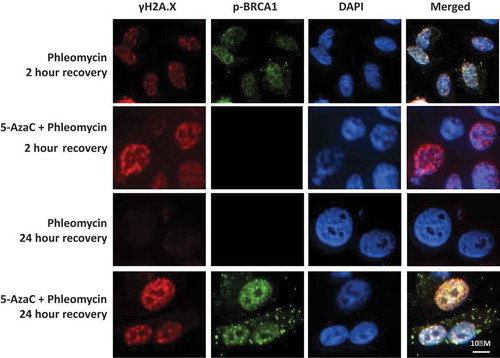
The delayed resolution of γH2A.X foci observed in cells treated with 5-AzaC might indicate an impaired DNA repair response. Previously we have shown that BRCA1 is transcriptionally up-regulated following a single dose of RT in PC-3 cells [Citation13] therefore we investigated how BRCA1 recruitment may be influenced by DNA hypomethylation in the presence of 5-AzaC. BRCA1 phosphorylated at serine 1423 (p-BRCA1) was analysed as this modification occurs in response to DNA damage and has previously been associated with radiation sensitivity [Citation19]. PC-3 cells treated with phleomycin alone exhibited numerous γH2A.X foci at 2-h post-exposure, with nuclear p-BRCA1 also being detected in these cells (). Control cells had resolved γH2AX foci by 24 h and p-BRCA1 was not detected at this time point (). In contrast, cells that had been treated with 5-AzaC prior to phleomycin treatment had high levels of DSBs at 2 h as reflected by γH2A.X foci levels however the corresponding increase in p-BRCA1 was not evident. Cells treated with 5-AzaC failed to resolve the γH2A.X foci by 24 h compared to the control. While p-BRCA1 was not detected at 2 h, it was detected at 24 h in these cells as evidenced by the punctate staining in the nucleus and cytoplasm. These data demonstrate that 5-AzaC treatment is associated with a delay in p-BRCA1 recruitment to DSBs and a delayed recovery from DNA damage.
Figure 5. Influence of 5-AzaC on BRCA1 recruitment and DNA repair. PC-3 cells were grown on chamber slides, either pre-treated with 5-azacytidine or left untreated, then treated with phleomycin for 1 h and left to recover for 2 or 24 h. Cells were then fixed and probed for p-BRCA1 (ser1423) and γH2A.X via immunofluorescence staining.
Discussion
One of the great challenges in cancer treatment is the individual variation in response to therapy. Identifying the key drivers of treatment resistance is integral in addressing this problem and in tailoring treatment. It is becoming increasingly evident that the epigenetic profile of cancer cells can impact response to therapy (reviewed in [Citation20]). By understanding adaptations that enable cancer cell survival against various treatment modalities, we can provide insight into the mechanisms of treatment resistance. Using prostate cancer cells with varying sensitivities to radiation-induced DNA damage, we sought to investigate how basal DNA methylation might influence response to treatment and how DNA methylation may be altered following DNA damage.
The data presented here show that each prostate cancer cell line displayed a distinct basal methylation profile. The radioresistant PC-3 cells exhibited higher levels of overall DNA methylation. There is evidence that increased CpG methylation is correlated with radioresistance. For example, radiation resistant head and neck squamous cell carcinoma cell lines have increased CpG methylation [Citation21] which is in agreement with our findings. Supporting the translational relevance for these findings, Chen et al. [Citation21] also demonstrated that radiation resistant head and neck cancer patient samples had increased CpG hypermethylation. The increased DNA methylation observed in the PC-3 cells however, does not appear to influence susceptibility to DNA damage as PC-3 and LNCaP cells displayed similar levels of γH2A.X foci following phleomycin-induced DNA damage. In fact, the PC-3 cells showed slightly higher basal levels of DNA damage than the LNCaP cells, indicating that the level of initial DNA damage does not determine cell survival. Instead, the variation in sensitivity to DNA damage in these cells may potentially be attributed to the efficacy of the subsequent DNA repair process. Alternatively, increased levels of DSBs may enhance the development of therapy-resistant clones via increased mutations and rapid genetic evolution. Both of these potential mechanisms warrant further investigation. It should be noted that while this immunofluorescence data provide valuable insight into the levels of DSBs in each cell line, the resolution of γH2A.X staining does not reflect 100% of the DSBs within the cell.
The random and transient nature of DSB induction means it is difficult to pinpoint and capture their corresponding epigenetic impact, here the coverage enabled by the Illumina Infinium HM450K array platform has provided insight on a finer scale. DNA damage induced by RT occurs at a higher frequency in regions of euchromatin and actively transcribed regions [Citation22,Citation23]. The HM450K probes are biased towards active promoter regions and these potential DNA damage hotspots. Overall, while very few changes in methylation were detected in response to RT, these indicate that subtle heritable changes in the epigenetic profile of cells exposed to DNA damaging agents are possible and may correlate with the sites of DSB repair. A limited number of localized DNA methylation changes are more likely than large genome-wide changes due to localized repair activity resulting in modifications to the site immediately surrounding the repaired DSB. A dose of 1 Gy would be expected to generate approximately 40 DSB lesions per cell [Citation24]. Our data identified between 33 and 39 CpG sites altered in the three cell lines following a dose of 2 Gy which represents about half the expected number of DSBs. However, the HM450K arrays cover less than 2% of all CpGs so there are many potential-impacted sites undetected in this analysis. A similar study by Antwih et al. [Citation25] investigated methylation changes in MDA-MB-231 breast cancer cells before and after RT. They demonstrated hundreds of genes which were hyper or hypomethylated following RT and a subset of these were functionally relevant, being involved in DNA repair, apoptosis and cell cycle regulation.
Numerous CpG sites displayed altered β values up to 14 d following exposure to 2 Gy, providing evidence for stable changes to DNA methylation following a single dose of RT. This is supported by the findings of O’Hagan et al. [Citation4], with DSB repair in an exogenous promoter model revealing long-term heritable gene silencing and indicating an ‘epigenetic memory’ of the DSB event and its subsequent repair. Similarly, repair of I-SceI restriction enzyme-induced DSBs driven by homologous recombination has been shown to trigger increased DNA methylation [Citation6]. Our data provide evidence for stable alterations to the epigenome in response to a single dose of RT which could potentially influence future response to treatment. The cumulative impact of localized DNA methylation changes following repeated DNA damage therefore needs to be determined. Ideally, to further characterize epigenetic adaptations to DNA damage, future work should focus on inducing DSBs at specific sites (perhaps using CRISPR/Cas9) within a chromatin context prior to DNA methylation profiling along with investigating other epigenetic alterations such as histone modifications and miRNAs.
The CpG sites identified as having altered methylation following RT may hold some predictive value, in fact ZMIZ1 promoter methylation has been patented for treatment selection and predicting improved patient survival in a range of cancers [Citation26]., with reduced ZMIZ1 expression associated with improved patient outcomes. In addition, HOXA11 methylation has been used to predict survival outcomes for AML patients [Citation14] and identified as a potential prognostic biomarker for ovarian cancer patients, with HOXA11 promoter methylation predicting poor outcomes in ovarian cancer [Citation15]. Reduced expression of HOXA11 has also been linked to treatment resistance in glioblastoma, including RT [Citation27]. These two CpGs located within the HOXA11 and ZMIZ1 gene bodies appear to be susceptible to DNA methylation changes in more than one cell line, indicating that they may be hotspots for DNA damage.
The DNA methylation inhibitor, 5-AzaC has shown promise as a DNA damage sensitizing agent, however the numerous cellular impacts of this DNA methylation inhibitor complicate and confound results. Pre-treating PC-3 cells with 5-AzaC did not sensitize them to DNA damage as some previous studies have shown [Citation9,Citation10,Citation17]. This may be due to the cellular context and differences in the inherent methylation profiles between different cell lines. In addition, sensitization by 5-AzaC may be modulated via its effects on transcription, RNA methylation (reviewed in [Citation28]), DSB induction [Citation29,Citation30], histone modifications [Citation31,Citation32] or via triggering interferon and viral defence pathways [Citation33]. It can also impact the localized recruitment of DNMT1 which has been shown to be recruited to sites of DNA repair [Citation34]. These factors could all potentially influence the DNA damage response and play a further confounding role in sensitization, demonstrating that the role for these inhibitors is certainly not clear cut.
While 5-AzaC did not sensitize prostate cancer cells to DNA damage, our immunofluorescence data demonstrated that 5-AzaC treatment delays recovery from DNA damage. Stalled DNA repair could potentially be attributed to the decrease in DNA methylation influencing signalling events required for recruiting DNA repair machinery or perhaps altered transcriptional programs are defective in managing protein levels required for the DNA damage response. DNMT1 trapping by 5-AzaC treatment may also be a key factor for this delay in DNA repair. A reduction in DNMT1 activity has previously been shown to inhibit the rate of DNA repair and 5-AzaC treatment has been shown to prevent DNMT1 recruitment to DSBs [Citation18]. Alternatively, the delay in recovery from DNA damage may be attributed to another cellular process influenced by 5-AzaC as mentioned previously. Our immunofluorescence data also revealed diffuse staining of DNMT1 in the absence of DNA damage, with more punctate staining evident in cells involved in repairing DSBs. This observation adds further support for the role of DNMT1 in DNA repair [Citation4,Citation18,Citation34].
In summary, our data provide evidence for stable epigenetic changes from a single dose of RT which has significant clinical implications in terms of cumulative effects of fractionated dosing regimens. While DNA methylation profiles in prostate cancer cells with divergent response to RT were stable on a genome-wide scale following a single dose of RT, a small subset of genes displayed altered CpG methylation following treatment. The sites identified exhibited a tendency towards gaining methylation and may represent regions of DSB repair. The random and transient nature of DSB induction and repair makes studying the epigenetic status at specific sites difficult however the advent of methylation arrays has enabled snapshots to be captured. Our data demonstrate that prostate cancer cells with a varied response to DNA damage have distinctive DNA methylomes. However, while the resistant cells generally exhibited higher levels of DNA methylation this did not influence susceptibility to DNA damage or the rate of short-term repair. In terms of clinical relevance, DNA methylation may have the potential to serve as a powerful biomarker of therapeutic response however the complexity of DNA repair within a chromatin context must be resolved. These heritable changes have the potential to influence not only the transcriptional profile of the cell but also future response to treatment. Clearly defining the interactions between the epigenome and DNA repair will only prove to strengthen our understanding of an individual’s tumour sensitivity and treatment efficacy.
Materials and methods
Cell culture and treatments
LNCaP and 22Rv1 cells were cultured in RPMI medium while PC-3 cells were cultured in HAMSF12K. All media were supplemented with 10% FBS (Bovogen SFBSA3) and 5000 U/ml Penicillin and 5000 μg/ml Streptomycin (Gibco 15070063). Cells were irradiated as previously described [Citation13]. The radiomimetic, phleomycin (Sapphire Biosciences 15549) was used at a final concentration of 50μg/ml for 1 h, before medium was removed, washed with PBS and replaced with fresh medium. Cells were then left to recover for the indicated time points. Cells were treated with 0.3 μM 5-azacytidine (5-AzaC, Sigma Aldrich A2385) or vehicle for 24 h, following which medium was aspirated and replaced with fresh medium. Cells were left to divide for a further 24 h before inducing DNA damage.
Illumina HM450K array sample preparation
DNA was extracted from cells recovering from 2 Gy irradiation at indicated time points using the Qiagen DNeasy kit. Illumina HM450K BeadChips were performed by the Australian Genome Research Facility.
Illumina HM450K array data processing
Raw signal intensity was imported into R (v 3.2) and processed using the minfi R package. Raw data underwent the standard Illumina normalization and SWAN [Citation35]. Individual probes with detection p-value higher than 0.05 were considered unreliable and removed from the analysis. In total, β and M values were calculated from 484,330 probes. To examine overall methylation similarities and dissimilarities, a density plot was generated using β-value and a hierarchical clustering using M-value. Heatmaps were generated using Morpheus (https://software.broadinstitute.org/morpheus/) based on β values of differentially methylated CpGs.
Bisulphite sequencing
Bisulphite sequencing was carried out as previously described [Citation36] using HOXA11 primers For: GAAAGTAGTTAAGTGGGGGTTGT, Rev: AACCAAAACTCAAACTAACAAAAAC.
Immunofluorescence assay
Cells were seeded onto Lab-Tek II eight-well chamber slides (Thermo Fisher Scientific) and treated with the appropriate agents before washing in warm PBS. Cells were then fixed using 4% Paraformaldehyde (Life Technologies 28906) for 10 min at room temperature. Fixed cells were then washed in PBS and permeabilized in 0.1% Triton-X. Cells were washed in PBS prior to blocking for 1 h at room temperature with shaking in blocking buffer (0.1% Tween-20 + 5% FCS) then incubated with the appropriate primary antibody (DNMT1 (SigmaAldrich D4567) used 1:100, BRCA1 phospho S1423 (Abcam ab47325) used at 1 μg/ml and γH2A.X [3F2] (Abcam ab22551) used at 3 μg/ml) overnight at 4°C. After washing each chamber in blocking buffer, the corresponding secondary antibody was added, the chamber slide wrapped in foil and incubated at room temperature for 1 h. Each well was then washed with PBS. The chamber apparatus was removed and DAPI/Fluroshield (Abcam ab104139) was added to the slide. After coverslipping and sealing, slides were stored in the dark at 4°C prior to imaging. Images were captured on the Andor Spinning Disk Confocal Microscope. FITC was captured at an exposure time of 200 ms, Alexa Fluor at 100 ms and DAPI at 100 ms.
Image analysis in Fiji Image Analysis software
Immunofluorescence images were analysed using Fiji Image Analysis software. Cells were isolated by first making the image binary and then using the find maxima function to localize the foci. Integrated density analysis was used to determine the number of foci for each cell. Results were generated in a table and copied to Microsoft Excel. After performing counts for 100 cells, the number of foci per cell was averaged and graphed in GraphPad Prism (Prism version 5.03 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com). P-values were calculated via Student’s t-test to determine statistical significance of differences between cell lines. One-way repeated measures ANOVA and Tukey’s post-test were used to determine statistical significance between treatments.
Clonogenic assay
Clonogenic assays were performed as previously described [Citation13]. Briefly, PC-3 cells were seeded at 1 × 103 cells per well and either left untreated or treated with 5-AzaC before phleomycin treatment at 1, 10, 50 or 100 μg/ml. After 14 d, colonies were fixed with 3:1 methanol to glacial acetic acid for 5 min. Following this, the fixative agent was removed, wells air dried completely and cells were stained with 1.0% methylene blue (Sigma-Aldrich, USA) in 50% ethanol for 30 min. Percentage of cell survival was calculated as the number of colonies post-treatment relative to the number of colonies within the corresponding control.
Annexin and propidium iodide staining
Apoptosis and necrosis were detected using the Alexa Fluor® 488 annexin V/Dead Cell Apoptosis Kit (Invitrogen, USA). Cells were analysed by flow cytometry using the FACSCantoTM Flow Cytometer (BD Biosciences, USA). PMT voltages were adjusted so negative control cells were within the first two log decades of the fluorescent parameter. Positively stained cell populations were discriminated on the basis of Alexa Fluor® 488 annexin V and PI control samples.
Author contributions
LPS, SAJ, JLP, AY, JEJ, RB and KHB performed experiments/analysis. MS, MA, PCT and AFH contributed to project design and manuscript preparation. KHB conceived and wrote manuscript.
Supplemental Material
Download MS Power Point (232.3 KB)Acknowledgments
The authors would like to thank the staff at the Holman Clinic at the Royal Hobart Hospital for cell irradiations.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015 Nov 19;60(4):547–560. PubMed PMID: 26590714; eng.
- Cann KL, Dellaire G. Heterochromatin and the DNA damage response: the need to relax. Biochem Cell Biol. 2011 Feb;89(1):45–60. PubMed PMID: 21326362; eng.
- Lorat Y, Schanz S, Schuler N, et al. Beyond repair foci: DNA double-strand break repair in euchromatic and heterochromatic compartments analyzed by transmission electron microscopy. PLoS One. 2012;7(5):e38165. PubMed PMID: 22666473; PubMed Central PMCID: PMC3364237.
- O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG Island. PLoS Genet. 2008 Aug;4(8). PubMed PMID: 18704159; PubMed Central PMCID: PMC2491723. eng. DOI:10.1371/journal.pgen.1000155
- Russo G, Landi R, Pezone A, et al. DNA damage and repair modify DNA methylation and chromatin domain of the targeted locus: mechanism of allele methylation polymorphism. Sci Rep. 2016 Sep 15;6:33222. PubMed PMID: 27629060; PubMed Central PMCID: PMC5024116. eng.
- Cuozzo C, Porcellini A, Angrisano T, et al. DNA damage, homology-directed repair, and DNA methylation. PLoS Genet. 2007 Jul;3(7):e110. PubMed PMID: 17616978; PubMed Central PMCID: PMC1913100. eng.
- Rogakou EP, Pilch DR, Orr AH, et al. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998 Mar 6;273(10):5858–5868. PubMed PMID: 9488723; Eng.
- Qiu H, Yashiro M, Shinto O, et al. DNA methyltransferase inhibitor 5-aza-CdR enhances the radiosensitivity of gastric cancer cells. Cancer Sci. 2009 Jan;100(1):181–188. PubMed PMID: 19037991.
- Hofstetter B, Niemierko A, Forrer C, et al. Impact of genomic methylation on radiation sensitivity of colorectal carcinoma. Int J Radiat Oncol Biol Phys. 2010 Apr;76(5):1512–1519. PubMed PMID: 20338477.
- Jiang W, Li YQ, Liu N, et al. 5-Azacytidine enhances the radiosensitivity of CNE2 and SUNE1 cells in vitro and in vivo possibly by altering DNA methylation. PLoS One. 2014;9(4). PubMed PMID: 24691157; PubMed Central PMCID: PMC3972231. eng. DOI:10.1371/journal.pone.0093273
- Kim HJ, Kim JH, Chie EK, et al. DNMT (DNA methyltransferase) inhibitors radiosensitize human cancer cells by suppressing DNA repair activity. Radiat Oncol. 2012;7:39. PubMed PMID: 22429326; PubMed Central PMCID: PMC3375186.
- Dote H, Cerna D, Burgan WE, et al. Enhancement of in vitro and in vivo tumor cell radiosensitivity by the DNA methylation inhibitor zebularine. Clin Cancer Res. 2005 Jun 15;11(12):4571–4579. PubMed PMID: 15958643.
- Young A, Berry R, Holloway AF, et al. RNA-seq profiling of a radiation resistant and radiation sensitive prostate cancer cell line highlights opposing regulation of DNA repair and targets for radiosensitization. BMC Cancer. 2014 Nov 4;14:808. PubMed PMID: 25369795; PubMed Central PMCID: PMC4233036.
- Bullinger L, Ehrich M, Dohner K, et al. Quantitative DNA methylation predicts survival in adult acute myeloid leukemia. Blood. 2010 Jan 21;115(3):636–642. PubMed PMID: 19903898; eng.
- Fiegl H, Windbichler G, Mueller-Holzner E, et al. HOXA11 DNA methylation – a novel prognostic biomarker in ovarian cancer. Int J Cancer. 2008 Aug 1;123(3):725–729. PubMed PMID: 18478570; eng.
- Fox KR, Grigg GW, Waring MJ. Sequence-selective binding of phleomycin to DNA. Biochem J. 1987 May 1;243(3):847–851. PubMed PMID: 2444208; PubMed Central PMCID: PMC1147934. eng.
- Brieger J, Mann SA, Pongsapich W, et al. Pharmacological genome demethylation increases radiosensitivity of head and neck squamous carcinoma cells. Int J Mol Med. 2012 Mar;29(3):505–509. PubMed PMID: 22109647; eng.
- Ha K, Lee GE, Palii SS, et al. Rapid and transient recruitment of DNMT1 to DNA double-strand breaks is mediated by its interaction with multiple components of the DNA damage response machinery. Hum Mol Genet. 2011 Jan 1;20(1):126–140. PubMed PMID: 20940144; PubMed Central PMCID: PMC3000680. Eng.
- Cortez D, Wang Y, Qin J, et al. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science (New York, NY). 1999 Nov 5;286(5442):1162–1166. PubMed PMID: 10550055; eng.
- Chi HC, Tsai CY, Tsai MM, et al. Impact of DNA and RNA methylation on radiobiology and cancer progression. Int J Mol Sci. 2018 Feb;19(2):555. PubMed PMID: 29439529; PubMed Central PMCID: PMC5855777. eng.
- Chen X, Liu L, Mims J, et al. Analysis of DNA methylation and gene expression in radiation-resistant head and neck tumors. Epigenetics. 2015 May 3;10(6):545–561. PubMed PMID: PMC4622425.
- Seo J, Kim SC, Lee H-S, et al. Genome-wide profiles of H2AX and γ-H2AX differentiate endogenous and exogenous DNA damage hotspots in human cells. Nucleic Acids Res. 2012 Mar 29;2012. DOI:10.1093/nar/gks287
- Takata H, Hanafusa T, Mori T, et al. Chromatin compaction protects genomic DNA from radiation damage. PLoS One. 2013;8(10):e75622. PubMed PMID: 24130727; PubMed Central PMCID: PMC3794047. eng.
- Mori R, Matsuya Y, Yoshii Y, et al. Estimation of the radiation-induced DNA double-strand breaks number by considering cell cycle and absorbed dose per cell nucleus. J Radiat Res. 2018 May;59(3):253–260. PubMed PMID: 29800455; PubMed Central PMCID: PMC5967466. eng.
- Antwih DA, Gabbara KM, Lancaster WD, et al. Radiation-induced epigenetic DNA methylation modification of radiation-response pathways. Epigenetics. 2013 Aug;8(8):839–848. PubMed PMID: 23880508; PubMed Central PMCID: PMC3883787.
- Mathios D, Lim M, Ha P, et al., inventor; Justia, assignee. Use of the ZMIZ1 marker in directing treatment and predicting survival in cancer. United States patent 20160273050. 2016 Mar 16.
- Se YB, Kim SH, Kim JY, et al. Underexpression of HOXA11 is associated with treatment resistance and poor prognosis in glioblastoma. Cancer Res Treat. 2017 Apr;49(2):387–398. PubMed PMID: 27456940; PubMed Central PMCID: PMC5398402. eng.
- Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008 Jul 1;123(1):8–13. PubMed PMID: 18425818; eng.
- Palii SS, Van Emburgh BO, Sankpal UT, et al. DNA methylation inhibitor 5-Aza-2ʹ-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol. 2008 Jan;28(2):752–771. PubMed PMID: 17991895; PubMed Central PMCID: PMC2223421. Eng.
- Kiziltepe T, Hideshima T, Catley L, et al. 5-Azacytidine, a DNA methyltransferase inhibitor, induces ATR-mediated DNA double-strand break responses, apoptosis, and synergistic cytotoxicity with doxorubicin and bortezomib against multiple myeloma cells. Mol Cancer Ther. 2007 Jun;6(6):1718–1727. PubMed PMID: 17575103; eng.
- Komashko VM, Farnham PJ. 5-azacytidine treatment reorganizes genomic histone modification patterns. Epigenetics. 2010 Apr;5(3):229–240. PubMed PMID: 20305384; eng.
- Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, et al. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci U S A. 2014 Jun 24;111(25):9169–9174. PubMed PMID: 24927542; PubMed Central PMCID: PMC4078803. Eng.
- Chiappinelli Katherine B, Strissel Pamela L, Desrichard A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015 Aug 27;162(5):974–986.
- Mortusewicz O, Schermelleh L, Walter J, et al. Recruitment of DNA methyltransferase I to DNA repair sites. Proc Natl Acad Sci U S A. 2005 Jun 21;102(25):8905–8909. PubMed PMID: 15956212; PubMed Central PMCID: PMC1157029.
- Maksimovic J, Gordon L, Oshlack A. SWAN: subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012 Jun 15;13(6):R44. PubMed PMID: 22703947; PubMed Central PMCID: PMC3446316. eng.
- Phillips JL, Taberlay PC, Woodworth AM, et al. Distinct mechanisms of regulation of the ITGA6 and ITGB4 genes by RUNX1 in myeloid cells. J Cell Physiol. 2018 Apr;233(4):3439–3453. PubMed PMID: 28926098; eng.