
ABSTRACT
Objectives
Describe the development of warm autoimmune hemolytic anemia warm (AIHA) secondary to a brown recluse spider (Loxosceles reclusa) bite is known as systemic loxoscelism; and review epidemiology, clinical manifestations, diagnostic work-up, pathophysiology, and treatment options associated with warm AIHA secondary to systemic loxoscelism.
Methods
Cases series of two cases of warm AIHA due to systemic loxoscelism and a review of the current literature: epidemiology, clinical manifestations, diagnostic work-up, pathophysiology, and treatment options associated with warm AIHA secondary to systemic loxoscelism.
Results
Presented here are two cases of warm AIHA due to systemic loxoscelism. Each patient was generally healthy appearing and presented with symptomatic anemia in the setting of brown recluse spider bites. Both patients were eventually found to have warm AIHA. Upon recognition of the diagnosis, the patients were started on corticosteroids and aggressive intravenous fluid hydration. In addition, they received transfusions of packed red blood cells. Their clinical courses improved, and they recovered to eventually be discharged home.
Conclusion
Envenomation by a brown recluse spider, Loxosceles reclusa, can result in systemic loxoscelism which can cause warm AIHA. The diagnosis of warm AIHA is confirmed by the direct antiglobulin/Coomb's test. Warm AIHA can be a life-threatening disease process. Hemodynamic support with intravenous fluids and RBC transfusion is the initial step in the management of these patients. Corticosteroids are the mainstay of current management. Second line treatments include rituximab. Rarely patients require splenectomy for refractory disease. Corticosteroids should be tapered over a three-month period.
Case report #1: A 30-year-old African American male with a necrotic lesion on upper extremity and hemolytic anemia
A 30-year-old African American male custodian with no significant past medical history presented for evaluation of nausea, myalgias, and an erythematous, eschar-like lesion on the patients left deltoid region (). He had been well until approximately 1 week prior to this admission when he noticed a painful, swollen, and erythematous lesion on his left shoulder. He denied any antecedent trauma. He denied fevers, chills, night sweats, or weight loss. He subsequently developed nausea and severe myalgias. He presented to the emergency department and was given intravenous fluids and discharged home on trimethoprim/sulfamethoxazole 160/800 mg by mouth twice daily.
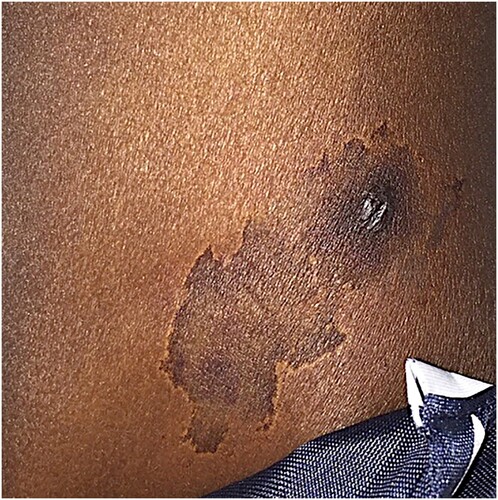
Figure 1. Skin manifestation of a 30-year-old African American male that presented with lesion on upper extremity and hemolytic anemia.
Over the following week he reported increasing generalized myalgia and weakness with associated nausea and non-bloody, bilious emesis. He denied diarrhea, constipation, abdominal pain, hematochezia, hematemesis, and melena. He denied palpitations, chest pain, orthopnea, and paroxysmal nocturnal dyspnea. He did report a mild degree of dyspnea on exertion.
He subsequently returned to the emergency department for reevaluation. Upon presentation, he was afebrile with a temperature of 99.8°F and hemodynamically stable with blood pressure of 121/59 mm of mercury, heart rate of 89 beats/minute, and respiratory rate of 20 breaths/minute. Physical examination revealed a well-developed 30-year-old African American male in no acute distress. He was noted to have bilateral scleral icterus, dry mucous membranes, mucosal pallor, no hepatosplenomegaly, a 1 cm raised, dark eschar on his left deltoid with a 1-inch surrounding area of hyperpigmentation (). The remainder of the physical examination was unremarkable.
Laboratory evaluation revealed a white blood cell count of 49.4 × 109/L, hemoglobin level of 4.4 g/dL, mean corpuscular hemoglobin concentration 31.2 g/dL, red blood cell distribution width 15.4%, blood urea nitrogen 37 mg/dL, calcium 8.3 mg/dL, bilirubin 8.97 mg/dL, aspartate aminotransferase 79 U/L, lactate dehydrogenase 1687 U/L, creatine kinase 621 U/L, and haptoglobin <10 mg/dL (). A direct antiglobulin test (DAT, also known as Coomb’s test) was obtained and revealed a positive result for both complement factor 3 and immunoglobulin G ().
Table 1. Case 1 Laboratory data from admission to discharge (Dr. Dickey case).
Based on the results of the DAT he was felt to have warm AIHA. He was started on methyl-prednisolone sodium succinate 500 mg intravenously every 12 h. Additionally, he received aggressive intravenous fluids were to decrease the risk of developing nephrotoxic acute tubular necrosis secondary to free hemoglobin. Lastly, he received transfusions of four units of packed red blood cells throughout his hospital stay.
His clinical course was uncomplicated and recovered without incident. He was discharged home on hospital day eight. At discharge, his corticosteroids were transitioned to prednisone 60 mg by mouth daily to be tapered over a 4-week period.
Case report #2: A 28-year-old African American female with a suspected spider bite and severe hemolytic anemia
A 28-year-old African American female with a past medical history of poly-substance abuse who presented for evaluation of back pain. She reported that she was in her usual state of health until approximately one week prior to admission when she noted low back pain. Her low back pain progressed to the point that she had difficulty ambulating. She denied fever, chills, night sweats, weight loss, palpitations, chest pain, orthopnea, paroxysmal nocturnal dyspnea, dyspnea on exertion, nausea, vomiting, diarrhea, constipation, abdominal pain, hematochezia, hematemesis, and melena.
She subsequently presented to the emergency department for evaluation. Upon presentation, she was afebrile with a temperature of 99.5°F, and hemodynamically stable with a blood pressure of 123/73 mm of mercury, heart rate of 98 beats/minute, and respiratory rate of 22 breaths/minute. Physical examination revealed a well-developed 28-year old African American female in mild distress. She was noted to have bilateral scleral icterus, moist mucous membranes, abdomen was soft, non-tender, and non-distended without masses or hepatosplenomegaly, and a 5 cm target-like lesion noted on her left upper back with central bulla and without eschar that was tender to palpation.
Laboratory evaluation revealed a white blood cell count of 34.5 × 109/L, hemoglobin level of 5.9 g/dL, mean corpuscular hemoglobin concentration 35 g/dL, red blood cell distribution width 16.8%, blood urea nitrogen 6.4 mg/dL, calcium 7.8 mg/dL, bilirubin 8.4 mg/dL, aspartate aminotransferase 925 U/L, lactate dehydrogenase 3247 U/L, creatine kinase not performed, and haptoglobin 10 mg/dL (). A direct antiglobulin test (DAT, also known as Coomb’s test) was obtained and revealed a positive result for both complement factor 3 and immunoglobulin G ().
Table 2. Case 2 Laboratory data from admission to discharge (Moore case).
Abdominal ultrasound also revealed an irregular-shaped gestational sac in the endometrial cavity. Her follow-up β-hCG level was ∼13,000 mLU/mL, indicating a fetus with gestational age of 6 weeks. Unfortunately, she developed intrauterine demise with incomplete spontaneous abortion. She subsequently underwent a dilation and curettage.
Based on the results of the DAT she was felt to have warm AIHA. She was started on methyl-prednisolone sodium succinate 500 mg intravenously every 12 h. Additionally, she received aggressive intravenous fluids were to decrease the risk of developing nephrotoxic acute tubular necrosis secondary to free hemoglobin. Lastly, she received transfusions of 5 units of packed red blood cells throughout her hospital stay.
On hospital day three, methyl-prednisolone sodium succinate was decreased to 250 mg intravenously every 12 h. On hospital day seven methyl-prednisolone sodium succinate was decreased to 75 mg intravenously every 12 h. Her hospital course was complicated by acute metabolic-toxic encephalopathy, most likely secondary to alcohol dependence and substance abuse withdrawal; however, other possibilities include high-dose corticosteroids. She was discharged home on hospital day nine. At discharge, her corticosteroids were transitioned to prednisone 40 mg by mouth daily to be tapered over a 4-week period.
Discussion
Systemic loxoscelism: epidemiology
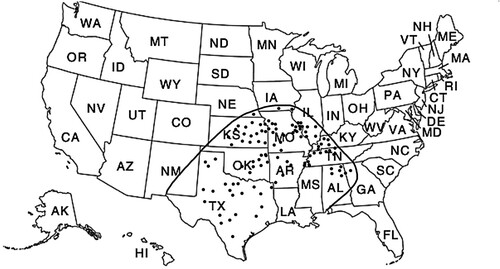
The brown recluse spider, Loxosceles reclusa, is typically characterized as a burnt orange to tan-colored arthropod, approximately 25 mm in diameter () [Citation1]. A characteristic feature of L. reclusa is a dark brown violin-shaped mark on the spider’s back () [Citation1]. L. reclusa is most commonly found within the Central Midwest to Southeastern and Southwestern regions of the U.S, with the highest population of L. reclusa located within Texas, Illinois, and Missouri () [Citation2]. According to the 2019 Annual Report of the American Association of Poison Control Centers, of the 4263 total spider bites, 802 were reported to do due to L. reclusa [Citation3]. Of the 802 envenomations, 148 suffered minor outcomes, 174 people suffered moderate outcomes, 24 suffered major outcomes, but no deaths were reported [Citation3]. However, Williams et al. concluded that from 1958 to 1995, approximately eight individual cases resulted in death due to systemic loxoscelism, likely secondary to acute kidney injury or disseminated intravascular coagulation [Citation4]. 17 cases of life-threatening L. reclusa induced hemolysis were reported between the years of 2003 and 2013, all of which occurred in children under the age of 18 and all in the Nashville, Tennessee area [Citation5].
Figure 2. A Brown Recluse spider (Loxosceles reclusa). Note the dark brown violin shaped image located on the spiders back. This can be utilized to identify L. reclusa.

Figure 3. Geographical distribution of the brown recluse spider (Loxosceles reclusa). Black dots represent L. reclusa populations. The highest density of L. reclusa is localized to Texas, Illinois, Missouri, Kansas, Oklahoma, and Kentucky.
Systemic loxoscelism: venom components and their pathological importance
Dermonecrosis
L. reclusa’s venom is a complex heterogenous mixture of sulfated nucleotide molecules, enzymatic compounds, and peptide toxins [Citation6,Citation7]. Chemical and experimental analysis of the venom reveals that the major compounds associated with local and systemic toxicity of L. reclusa envenomation are hyaluronidases, phospholipase D, sphingomyelinase D, collagenase, and phosphodiesterases [Citation6].
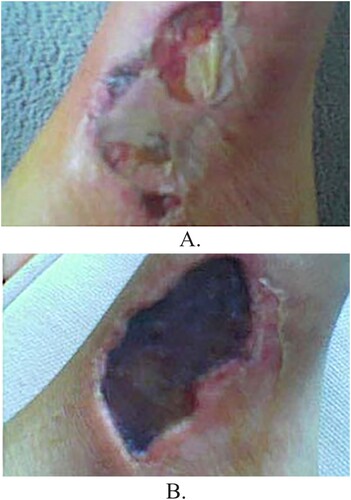
Initially, the bite is painless, and most victims are unaware of having been bitten. Within the first 10 min of envenomation local toxicity and subsequent inflammation occurs. Hyalurondiase and collagenase begin breaking down the extracellular matrix of the surrounding tissue. Additionally, sphingomyelinase and phospholipase begin cleaving cellular membrane phospholipids, thereby yielding sphingomyelin and arachidonic acid, respectfully [Citation6]. Arachidonic acid is then metabolized yielding prostaglandins (PG), thromboxane (TX), and leukotrienes (LT). These compounds, along with proinflammatory cytokines, begin to recruit granulocytes to the area of envenomation. Eventually, the combination of venom components, cellular death, and aggressive inflammatory responses lead to hemorrhagic necrosis and ultimately the formation of dermonecrosis (also known as necrotic arachnidism) ((B)). Necrotic arachnidism typically presents as a painful erythematous area that ulcerates and develops a central black eschar ((A,B) [Citation8].
L. reclusa induced hemolytic anemia
The exact mechanism of L. reclusa induced hemolytic anemia still remains unclear. One theory is that direct red blood cell (RBC) hemolysis is related to the compounds found within the venom of L. reclusa, particularly sphingomyelinase. In vitro studies have documented that sheep and human RBCs lyse when exposed to L. reclusa venom [Citation9]. Additionally, there is evidence for the direct attachment of venom compounds to human RBC membranes using a ferritin-labeled antibody technique [Citation10]. This attachment is dose dependent, which could potentially explain why children exhibit a more aggressive hemolytic reaction from L. reclusa envenomation [Citation10]. RBCs exposed to L. reclusa venom failed to undergo hemolysis when incubated with complement-depleted plasma in vitro [Citation5] This provides evidence that complement-mediated hemolysis is a key step in the pathogenesis of L. reclusa induced hemolytic anemia. There is also an indirect relationship between Eculizumab, a recombinant monoclonal C5 convertase inhibitor, and RBC hemolysis, again providing additional support for complement mediated hemolysis [Citation5]
Warm (IgG) Autoimmune Hemolytic Anemia (warm AIHA)
L. relusa induced hemolytic anemia can be extravascular, intravascular, or both [Citation11]. Multiple case reports have provided evidence to support the idea that immunoglobulin G (IgG) antibodies and complement activation are both involved in this mechanism, as indicated by a positive direct Coomb’s test (also known as direct antiglobulin test) [Citation4,Citation12]. A few case reports have found that despite a positive direct Coomb’s test, only complement factor C3 was present on RBCs but IgG antibodies were not [Citation13,Citation14] The findings in these case reports are supported by data confirming that complement activation in the absence of IgG antibodies revealed the presence of IgM suggesting IgM mediated complement activation [Citation15]
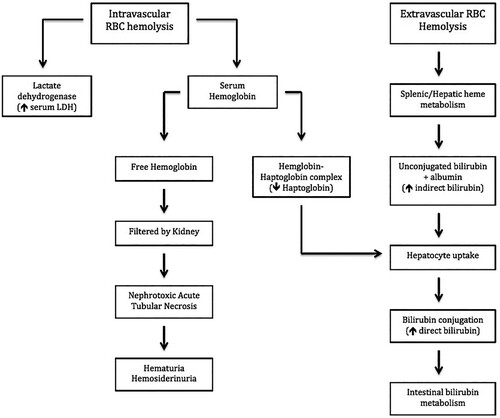
This demonstrates the role of complement activation leading to intravascular hemolysis. This in turn could set the stage for opsonization by splenic and hepatic macrophages via deposition of complement factor C3b on the RBCs membrane thereby mediating extravascular hemolysis (). Another study found, via flow cytometry, that the classic pathway components C1q, C3 and C4 were found deposited on RBC’s treated with sphingomyelinase toxins P1 and P2 from L. intermedia venom [Citation16]
Figure 6. Intravascular and extravascular hemolysis secondary to L. reclusa envenomation. Note that RBC hemolysis leads to the release of intracellular LDH thereby leading to an elevated serum LDH level. In addition, intravascular RBC hemolysis leads to the release of free hemoglobin which eventually binds serum haptoglobin thereby forming a haptoglobin-hemoglobin complex (Hap-Hem). The Hap-Hem complex results in the reduction of circulating free haptoglobin. Importantly, high concentrations of intraluminal hemoglobin within the kidney predisposes to nephrotoxic acute tubular necrosis. Therefore it is important to aggressive hydrate patients with severe hemolytic anemia, such as in systemic loxoscelism.
RBCs that were exposed to L. reclusa venom resulted in the deposition of both IgG and complement after incubation with ABO identical fresh frozen plasma [Citation5]. The addition of eculizumab, a recombinant monoclonal antibody that inhibits C5 convertase, reduced RBC hemolysis at 24 h of incubation from 10.9% hemolysis without eculizumab to nondetectable with Eculizumab (p < 0.001) [Citation5] Similar results were obtained at 48, 72, and 96 h [Citation5]. Overall, eculizumab reduced venom-mediated hemolysis by 79.2% [Citation5]. These results would suggest a possible emerging therapeutic option L. reclusa mediated hemolytic anemia.
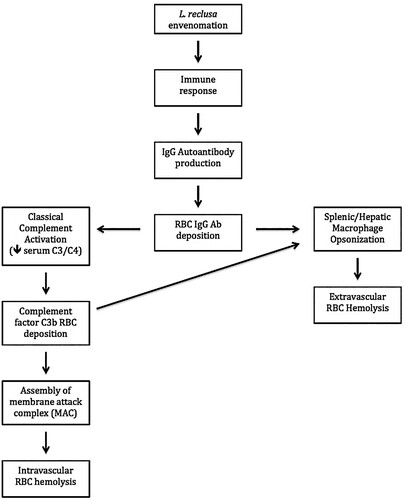
IgM antibodies are much more efficient at activating the classic complement pathway due to ability to bind complement component C1q. IgG subclasses are also capable of activating complement, although less effectively due to its smaller dimensions and reduced affinity for C1q [Citation17]. It is hypothesized that L. reclusa venom induces an immune response that results in the production of significant amounts of IgG autoantibodies (). These autoantibodies would bind to RBCs thereby activating the classical complement pathway (intravascular hemolysis pathway), and simultaneously inducing opsonization by splenic and hepatic macrophages (extravascular hemolysis pathway) ().
Figure 5. Hypothesized pathophysiological response to L. reclusa induced hemolytic anemia. Note that complement factor C3b and IgG deposit onto the RBC surface thereby initiating RBC phagocytosis. Importantly, the direct antiglobulin test (DAT or Coomb’s test) identifies the deposition of C3b and IgG deposition, which makes the DAT extremely valuable in detecting warm AIHA. In addition, activation of the classical complement pathway results in the assembly of C1 complex, C4, and C2 thereby forming the C3 convertase (C2aC4b), which leads to a reduction of circulating complement factors C3 and C4.
Systemic loxoscelism: diagnosis
History of presenting illness
A definitive diagnostic study for systemic loxoscelism does not exist. A few publications have utilized a skin surface ELISA technique in order to identify L. reclusa venom [Citation18,Citation19]. 90% of envenomation’s by L. reclusa resolve without dermonecrosis or major complications, thus the utility of a L. reclusa specific ELISA is unknown [Citation20]. Systemic loxoscelism is primarily a clinical diagnosis. The presenting history, physical exam, and laboratory data are extremely important in recognizing systemic loxoscelism, as a delay in recognition, and thus treatment, can be fatal [Citation4]
The clinical course of systemic loxoscelism is unpredictable. Certain risk factors can place the patient in a higher risk group: African American and Hispanic ethnicity, children less than 18 years of old, and patients with underlying hemoglobinopathies, or intrinsic erythrocyte defects. Since systemic loxoscelism is a clinical diagnosis, the clinician should ascertain if the patient resides in, or has traveled to, an areae where L. reclusa is endemic (). The clinician should also provide the patient with visual representation of L. reclusa, as this could help identify the difference between L. reclusa and other arthropods (). Additional symptoms commonly found in patients with systemic loxoscelism include fever, malaise, abdominal pain, profound fatigue, nausea, vomiting, diarrhea, headache, and rarely delirium [Citation21] ().
Table 3. Signs and symptoms of systemic loxoscelism.
Physical exam
Typically, the site of envenomation becomes erythematous and warm within the first 24 h following the bite. Between 48 and 72 h, the site of envenomation develops into a bullous appearing erythematous lesion ((A)). The lesion then ulcerates to become necrotic and a central black eschar (dermonecrosis) begins to form [Citation18,Citation21] (). Other physical exam findings include pale conjunctiva, scleral icterus, jaundice, right upper quadrant pain, splenomegaly, petechiae [Citation21–24] urticaria [Citation4,Citation25–28], maculopapular rash [Citation4,Citation21,Citation29], dark brown urine [Citation4] tachypnea and tachycardia ().
Figure 4. (A) Purulent cutaneous lesion due to L. reclusa approximately 1 week after the initial bite. (B) Dermonecrosis due to L. reclusa 2 weeks after the initial bite. (Yigit). The combination of venom components, cellular death, and aggressive inflammatory response leads to hemorrhagic necrosis and ultimately the formation of dermonecrosis with a central black eschar.
Laboratory data
L. reclusa induced hemolytic anemia can occur as early as 24 h following, and almost always within the first 96 h of, envenomation [Citation21,Citation30]. L. relcusa induced hemolytic anemia is suspected to occur secondary to intravascular complement activation, as well as splenic and hepatic erythrocyte opsonization and phagocytosis. Findings associated with intravascular hemolysis include reduced hemoglobin and hematocrit, reduced or absent haptoglobin, elevated lactate dehydrogenase (LDH), elevated serum/urine free hemoglobin, and reduced levels of complement factors C3 and C4 (). Findings that are associated with extravascular hemolysis include elevated LDH, and direct [Citation21] or indirect [Citation4] hyperbilirubinemia ().
Table 4. Laboratory findings associated with systemic loxoscelism.
Some cases have reported reduced fibrinogen, elevated D-dimer [Citation30], prolonged prothrombin time and partial thromboplastin time, indicating disseminated intravascular coagulopathy [Citation4,Citation30–32].
Urinalysis demonstrates proteinuria, hemosiderinuria, increased urobilinogen, and hemoglobinuria [Citation4,Citation33] (). With severe hemolysis, high concentrations of free urine hemoglobin can predispose to nephrotoxic acute tubular necrosis [Citation21].
Additional tests that can help the clinician identify warm (IgG) AIHA are the direct antiglobulin test (DAT) (also known as a direct Coomb’s test) and a peripheral blood smear. Most cases of system loxoscelism have reported a positive DAT [Citation12], however, a few cases have reported a negative DAT [Citation4,Citation21] as well. The negative DAT in these particular cases could very well be false negatives due to IgM autoantibodies (which are not detected by the DAT test) or RBC-bound IgG below the threshold of the DAT test [Citation34]. Spherocytes on the peripheral blood smear should also direct the clinician towards warm (IgG) AIHA.
Diagnosis of warm AIHA is established when all of the following are present: (1) hemolytic anemia as demonstrated by anemia, high LDH, low or absent haptoglobin, and indirect hyperbilirubinemia (2) presence of spherocytes on peripheral blood smear and (3) a positive DAT/Coomb’s test ().
Table 5. Diagnosing warm (IgG) autoimmune hemolytic anemia.
Systemic loxoscelism: management
A definitive therapeutic approach to systemic loxoscelism has yet to be established in the medical literature, however, several interventions have been proposed. Initial management involves hemodynamic support with intravenous fluids and RBC transfusion. Aggressive intravenous fluids are necessary to decrease the development of nephrotoxic acute tubular necrosis secondary to free hemoglobin in the setting of hemolysis. Although, systemic corticosteroids have been shown to be effective [Citation23,Citation30,Citation32,Citation35,Citation36], it remains unclear if corticosteroids alone are adequate in severe cases of systemic loxoscelism. Recent publications have suggested that L. reclusa induced hemolytic anemia occurs secondary to the deposition of IgG autoantibodies. Thus, the treatment algorithm for systemic loxoscelism should therefore reflect the current treatment regimen for any cause of warm AIHA. Here we will provide additional support for corticosteroids in the treatment for systemic loxoscelism, as well as investigate additional pharmaceutical and surgical modalities, such as rituximab, splenectomy, intravenous immunoglobulin therapy (IVIG), eculizumab, and other monoclonal antibodies ( and ), all of which could possibly be utilized in patients that do not respond to initial management with systemic corticosteroids.
Table 6. Treatment modalities with response rate (RR) for systemic loxoscelism.
Table 7. Summary.
Dapsone
Based on the literature, dapsone has been recommended for cutaneous loxoscelism, but not currently advocated as monotherapy for systemic loxoscelism [Citation23]. The utilization of dapsone in patients with concurrent cutaneous and systemic loxoscelism should be thoroughly assessed, as dapsone itself can cause hemolytic anemia and methemoglobinemia, thereby potentially worsening the patient’s prognosis [Citation30]. Recent animal studies suggest that neither dapsone, nor the combination of dapsone plus hyperbaric oxygen, reduced dermonecrosis from L. reclusa envenomation [Citation36,Citation37]. As well, dapsone requires further testing for glucose 6-phosphate dehydrogenase (G6PD) activity prior to administration. The risk-to-benefit ratio of using dapsone is relatively modest.
Corticosteriods
Corticosteroids for systemic loxoscelism have not been subjected to clinical trials. Therefore, the exact dosage and duration of therapy have not been established. Additionally, risk factors that predispose the patient to a more severe hemolytic anemia have not been established. The utilization of corticosteroids for the treatment of systemic loxoscelism is common, but more data is needed to determine efficacy.
Corticosteroids are first-line treatment of warm (IgG) AIHA. The distinction between mild hemolytic anemia and severe hemolytic anemia is particularly vague, as this presents complications when determining the appropriate corticosteroid regimen. Patients with warm (IgG) AIHA should receive an initial dose of prednisone 1.0–2.0 mg/kg/day or the equivalent dose of methylprednisolone intravenously [Citation38]. These doses are continued until the hemoglobin is greater than 10 g/dL.
It is important that one not abruptly discontinue corticosteroids in the setting of AIHA. It is suggested that the patient be treated with low-dose prednisone (<10 mg/day) for a minimum of three months. This duration is thought to reduce the incidence of relapsed AIHA and prolong remission rates [Citation39]. It is estimated that the utilization of corticosteroids as the first-line therapy for warm (IgG) AIHA provides a therapeutic response in 70–85% of patients, however, only 33% of patients remain in long-term remission [Citation34].
Rituximab
Rituximab, a chimeric anti-CD20 monoclonal antibody, is used with, or without, corticosteroids for the treatment of warm AIHA [Citation40]. A recent publication concluded that patients with refractory warm AIHA had an overall response rate of 70–89% when treated either a low dose (100 mg IV weekly infusions for 4 weeks) or a maintenance dose of rituximab (375 mg IV weekly infusions for 4 weeks) [Citation41–49]. A 2015 meta-analysis of 21 studies encompassing 409 patients concluded that patients treated with rituximab had an overall response rate of 73% [Citation50]. The median time to response was 4–6 weeks with a range of 1 week to 3 months following the first dose of rituximab [Citation41]. The most common adverse events associated with rituximab appear to be infusion-related reactions, however, progressive multifocal encephalopathy, hepatitis B reactivation and viral infections have also been reported [Citation40,Citation42,Citation49,Citation52,Citation53]. Patients receiving rituximab should be premedicated with methylprednisolone 100 mg intravenously, chlorpheniramine 10 mg intravenously, and oral acetaminophen 1 gram 1 h prior to infusion to reduce infusion-related reactions. Rituximab seems to be a safe and effective alternative treatment for warm AIHA that could be utilized prior a more invasive therapy, such as surgery.
Splenectomy
Erythrocyte destruction associated with warm AIHA occurs mainly in the spleen. Therefore, splenectomy is an alternative treatment approach for warm AIHA. There are no clinical trials comparing splenectomy to other second-line therapeutic treatments of warm AIHA. A prospective study conducted by Akpek found that approximately 82% (9 or 11) of patients with refractory idiopathic warm AIHA had complete remission following splenectomy [Citation54]. Despite the relatively high remission rate, the risk of developing sepsis due to encapsulated bacteria is approximately 5% and carries a mortality rate of 50%, even with proper vaccinations [Citation55,Citation56] A study concluded that 11% of 308 patients with warm AIHA developed thrombotic events post-splenectomy [Citation52] Based on the high mortality rate and risk of thrombotic events post-splenectomy, it is suggested that splenectomy be reserved for refractory warm AIHA following the failure of first- and second-line therapeutic approaches.
Other treatment approaches: immunosuppressive agents and IVIG
Prior to second-line therapies such as rituximab, immunosuppressive agents like azathioprine, cyclophosphamide, cyclosporine A, and mycophenolate mofetil were used in multiple case reports, however, the response rates were relatively low (40–60%) compared to rituximab (70–89%) [Citation41–44,Citation46–49,Citation51,Citation57–64]. Given the relatively low response rate, in conjunction with the extensive adverse effects that can be associated with immunosuppressive agents, the risk to benefit ratio of these agents in the treatment of warm AIHA is markedly high.
Intravenous immunoglobulins (IVIG) use in the treatment of warm AIHA should only be used in the setting of rescue therapy for inadequate response to corticosteroids and/or rituximab per recent international consensus recommendations [Citation65]. A retrospective study of 73 patients with recurrent warm AIHA secondary to chronic lymphocytic leukemia reported a relatively fair response rate of 40% in adults and 54% in children following the use of IVIG [Citation66,Citation67]
Summary
Envenomation by a brown recluse spider, L. reclusa, can result in systemic loxoscelism which can cause warm AIHA. The diagnosis of warm AIHA is confirmed by the direct antiglobulin/Coomb’s test. Warm AIHA can be a life-threatening disease process and should be managed urgently/emergently. Hemodynamic support with intravenous fluids and RBC transfusion is the initial step in the management of these patients. Corticosteroids are the mainstay of current management. The first-line treatment for warm AIHA is corticosteroids, which has a response rate of approximately 70–85%. Second line treatments include rituximab (response rate: 73%). Rarely patients require splenectomy (response rate: 82%) for refractory disease. Corticosteroids should be tapered over a three-month period. The patient should follow-up with a hematologist/oncologist within one week after discharge and should be evaluated with a complete blood count, comprehensive metabolic panel, reticulocyte count, LDH, and haptoglobin level. In the event of refractory or relapsed warm autoimmune hemolytic anemia, the physician should consider either rituximab or splenectomy. In addition, the patient should maintain a tapering regimen of prednisone for 4–6 weeks.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Potter MF. Brown recluse spider. University of Kentucky College of Agriculture. ENTFACT-631. https://entomology.ca.uky.edu/ef631.
- Vetter RS. Arachnids submitted as suspected brown recluse spiders (Araneae: Sicariidae): Loxosceles spiders are virtually restricted to their known distributions but are perceived to exist throughout the United States. J Med Entomol. 2005;42(4):512–521. doi:10.1093/jmedent/42.4.512.
- Gummin DD, Mowry JB, Beuhler MC, et al. 2019 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 37th Annual Report. Clin Toxicol (Phila). 2020;58(12):1360–1541. doi:10.1080/15563650.2020.1834219.
- Williams ST, Khare VK, Johnston GA, et al. Severe intravascular hemolysis associated with brown recluse spider envenomation. A report of two cases and review of the literature. Am J Clin Pathol. 1995;104(4):463–467. doi:10.1093/ajcp/104.4.463.
- Gehrie EA, Nian H, Young PP. Brown recluse spider bite mediated hemolysis: clinical features, a possible role for complement inhibitor therapy, and reduced RBC surface glycophorin A as a potential biomarker of venom exposure. PLoS One. 2013;8(9):e76558. Published 2013 Sep 27. doi:10.1371/journal.pone.0076558.
- Sannaningaiah D, Subbaiah GK, Kempaiah K. Pharmacology of spider venom toxins. Toxin Rev. 2014;33(4):206–220. doi: 10.3109/15569543.2014.954134.
- Schroeder FC, Taggi AE, Gronquist M, et al. NMR-spectroscopic screening of spider venom reveals sulfated nucleosides as major components for the brown recluse and related species. Proc Natl Acad Sci U S A. 2008;105(38):14283–14287. doi:10.1073/pnas.0806840105.
- Yigit N, Bayram A, Ulasoglu D, et al. Loxosceles spider bite in Turkey (Loxosceles rufescens, Sicariidae, Araneae). J Venom Anim Toxins Incl Trop Dis. 2008;14(1):178–187.
- Forrester LJ, Barrett JT, Campbell BJ. Red blood cell lysis induced by the venom of the brown recluse spider: the role of sphingomyelinase D. Arch Biochem Biophys. 1978;187(2):355–365. doi:10.1016/0003-9861(78)90046-2.
- Futrell JM, Morgan PN, Su SP, et al. Location of brown recluse venom attachment sites on human erythrocytes by the firritin-labeled antibody technique. Am J Pathol. 1979;95(3):675–682.
- Loden JK, Seger DL, Spiller HA, et al. Cutaneous-hemolytic loxoscelism following brown recluse spider envenomation: new understandings. Clin Toxicol (Phila). 2020;58(12):1297–1305. doi:10.1080/15563650.2020.1739701.
- Eichner ER. Spider bite hemolytic anemia: positive Coombs’ test, erythrophagocytosis, and leukoerythroblastic smear. Am J Clin Pathol. 1984;81(5):683–687. doi:10.1093/ajcp/81.5.683.
- Lane DR, Youse JS. Coombs-positive hemolytic anemia secondary to brown recluse spider bite: a review of the literature and discussion of treatment. Cutis. 2004;74(6):341–347.
- McDade J, Aygun B, Ware RE. Brown recluse spider (Loxosceles reclusa) envenomation leading to acute hemolytic anemia in six adolescents. J Pediatr. 2010;156(1):155–157. doi:10.1016/j.jpeds.2009.07.021.
- Meulenbroek EM, de Haas M, Brouwer C, et al. Complement deposition in autoimmune hemolytic anemia is a footprint for difficult-to-detect IgM autoantibodies. Haematologica. 2015;100(11):1407–1414. doi:10.3324/haematol.2015.128991.
- Tambourgi DV, Da Silva M DS, Billington SJ, et al. Mechanism of induction of complement susceptibility of erythrocytes by spider and bacterial sphingomyelinases. Immunology. 2002;107(1):93–101. doi:10.1046/j.1365-2567.2002.01483.x.
- Coulie PG, Van Snick J. Enhancement of IgG anti-carrier responses by IgG2 anti-hapten antibodies in mice. Eur J Immunol. 1985;15(8):793–798. doi:10.1002/eji.1830150810.
- Stoecker WV, Wasserman GS, Calcara DA, et al. Systemic loxoscelism confirmation by bite-site skin surface: ELISA. Mo Med. 2009;106(6):425–431.
- McGlasson DL, Green JA, Stoecker WV, et al. Duration of Loxosceles reclusa venom detection by ELISA from swabs. Clin Lab Sci. 2009;22(4):216–222.
- King LE, Rees RS. Spider bites and scorpion stings. Curr Ther. 1978;1978:970–973.
- Michaud ME, Gibler WB. Hemolytic anemia and hemoglobinuria due to systemic loxoscelism: report of a case. J Wilderness Med. 1991;2(1):49–54.
- Hobbs GD, Jr HR. Brown recluse spider bites: a common cause of necrotic arachnidism. Am J Emerg Med. 1989;7(3):309–312. doi:10.1016/0735-6757(89)90178-2.
- Sams HH, Dunnick CA, Smith ML Jr, et al. Necrotic arachnidism. J Am Acad Dermatol. 2001;44(4):561–576. doi:10.1067/mjd.2001.112385.
- Lucas S. Spiders in Brazil. Toxicon. 1988;26(9):759–772. doi:10.1016/0041-0101(88)90317-0.
- Futrell JM. Loxoscelism. Am J Med Sci. 1992;304(4):261–267. doi:10.1097/00000441-199210000-00008.
- Wilson DC, King LE Jr. Spiders and spider bites. Dermatol Clin. 1990;8(2):277–286.
- Cardoso JL, Wen FH, França FO, et al. Detection by enzyme immunoassay of Loxosceles gaucho venom in necrotic skin lesions caused by spider bites in Brazil. Trans R Soc Trop Med Hyg. 1990;84(4):608–609. doi:10.1016/0035-9203(90)90058-m.
- Gendron BP. Loxosceles reclusa envenomation. Am J Emerg Med. 1990;8(1):51–54. doi:10.1016/0735-6757(90)90297-d.
- Lane L, McCoppin HH, Dyer J. Acute generalized exanthematous pustulosis and Coombs-positive hemolytic anemia in a child following Loxosceles reclusa envenomation. Pediatr Dermatol. 2011;28(6):685–688. doi:10.1111/j.1525-1470.2010.01302.x.
- Wasserman GS, Anderson PC. Loxoscelism and necrotic arachnidism. J Toxicol Clin Toxicol. 1983;21(4-5):451–472. doi:10.3109/15563658308990434.
- Vorse H, Seccareccio P, Woodruff K, et al. Disseminated intravascular coagulopathy following fatal brown spider bite (necrotic arachnidism). J Pediatr. 1972;80(6):1035–1037. doi:10.1016/s0022-3476(72)80023-4.
- Hogan CJ, Barbaro KC, Winkel K. Loxoscelism: old obstacles, new directions. Ann Emerg Med. 2004;44(6):608–624. doi:10.1016/j.annemergmed.2004.08.028.
- Sezerino UM, Zannin M, Coelho LK, et al. A clinical and epidemiological study of Loxosceles spider envenoming in Santa Catarina, Brazil. Trans R Soc Trop Med Hyg. 1998;92(5):546–548. doi:10.1016/s0035-9203(98)90909-9.
- Zanella A, Barcellini W. Treatment of autoimmune hemolytic anemias. Haematologica. 2014;99(10):1547–1554. doi:10.3324/haematol.2014.114561.
- Anderson PC. Spider bites in the United States. Dermatol Clin. 1997;15(2):307–311. doi:10.1016/s0733-8635(05)70438-1.
- Hobbs GD, Anderson AR, Greene TJ, et al. Comparison of hyperbaric oxygen and dapsone therapy for loxosceles envenomation. Acad Emerg Med. 1996;3(8):758–761. doi:10.1111/j.1553-2712.1996.tb03511.x.
- Phillips S, Kohn M, Baker D, et al. Therapy of brown spider envenomation: a controlled trial of hyperbaric oxygen, dapsone, and cyproheptadine. Ann Emerg Med. 1995;25(3):363–368. doi:10.1016/s0196-0644(95)70296-2.
- Brodsky RA. Warm autoimmune hemolytic anemia. N Engl J Med. 2019;381(7):647–654. doi:10.1056/NEJMcp1900554.
- Dussadee K, Taka O, Thedsawad A, et al. Incidence and risk factors of relapses in idiopathic autoimmune hemolytic anemia. J Med Assoc Thai. 2010;93(Suppl 1):S165–S170.
- Garvey B. Rituximab in the treatment of autoimmune haematological disorders. Br J Haematol. 2008;141(2):149–169. doi:10.1111/j.1365-2141.2008.07054.x.
- Dierickx D, Kentos A, Delannoy A. The role of rituximab in adults with warm antibody autoimmune hemolytic anemia. Blood. 2015;125(21):3223–3229. doi:10.1182/blood-2015-01-588392.
- Zecca M, Nobili B, Ramenghi U, et al. Rituximab for the treatment of refractory autoimmune hemolytic anemia in children. Blood. 2003;101(10):3857–3861. doi:10.1182/blood-2002-11-3547.
- Narat S, Gandla J, Hoffbrand AV, et al. Rituximab in the treatment of refractory autoimmune cytopenias in adults. Haematologica. 2005;90(9):1273–1274.
- D'Arena G, Califano C, Annunziata M, et al. Rituximab for warm-type idiopathic autoimmune hemolytic anemia: a retrospective study of 11 adult patients. Eur J Haematol. 2007;79(1):53–58. doi:10.1111/j.1600-0609.2007.00861.x.
- Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119(16):3691–3697. doi:10.1182/blood-2011-06-363556.
- Maung SW, Leahy M, O'Leary HM, et al. A multi-centre retrospective study of rituximab use in the treatment of relapsed or resistant warm autoimmune haemolytic anaemia. Br J Haematol. 2013;163(1):118–122. doi:10.1111/bjh.12486.
- Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163(3):393–399. doi:10.1111/bjh.12541.
- Roumier M, Loustau V, Guillaud C, et al. Characteristics and outcome of warm autoimmune hemolytic anemia in adults: New insights based on a single-center experience with 60 patients. Am J Hematol. 2014;89(9):E150–E155. doi:10.1002/ajh.23767.
- Bussone G, Ribeiro E, Dechartres A, et al. Efficacy and safety of rituximab in adults’ warm antibody autoimmune haemolytic anemia: retrospective analysis of 27 cases. Am J Hematol. 2009;84(3):153–157. doi:10.1002/ajh.21341.
- Reynaud Q, Durieu I, Dutertre M, et al. Efficacy and safety of rituximab in auto-immune hemolytic anemia: A meta-analysis of 21 studies. Autoimmun Rev. 2015;14(4):304–313. doi:10.1016/j.autrev.2014.11.014.
- Barcellini W, Zanella A. Rituximab therapy for autoimmune haematological diseases. Eur J Intern Med. 2011;22(3):220–229. doi:10.1016/j.ejim.2010.12.016.
- Barcellini W, Fattizzo B, Zaninoni A, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood. 2014;124(19):2930–2936. doi:10.1182/blood-2014-06-583021.
- Rao A, Kelly M, Musselman M, et al. Safety, efficacy, and immune reconstitution after rituximab therapy in pediatric patients with chronic or refractory hematologic autoimmune cytopenias. Pediatr Blood Cancer. 2008;50(4):822–825. doi:10.1002/pbc.21264.
- Akpek G, McAneny D, Weintraub L. Comparative response to splenectomy in Coombs-positive autoimmune hemolytic anemia with or without associated disease. Am J Hematol. 1999;61(2):98–102. doi:10.1002/(sici)1096-8652(199906)61:2<98::aid-ajh4>3.0.co;2-g
- Bisharat N, Omari H, Lavi I, et al. Risk of infection and death among post-splenectomy patients. J Infect. 2001;43(3):182–186. doi:10.1053/jinf.2001.0904.
- Davidson RN, Wall RA. Prevention and management of infections in patients without a spleen. Clin Microbiol Infect. 2001;7(12):657–660. doi:10.1046/j.1198-743x.2001.00355.x.
- Lechner K, Jäger U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116(11):1831–1838. doi:10.1182/blood-2010-03-259325.
- Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24(3):195–210. doi:10.1016/j.tmrv.2010.03.002.
- Alba P, Karim MY, Hunt BJ. Mycophenolate mofetil as a treatment for autoimmune haemolytic anaemia in patients with systemic lupus erythematosus and antiphospholipid syndrome. Lupus. 2003;12(8):633–635. doi:10.1191/0961203303lu419cr.
- Howard J, Hoffbrand AV, Prentice HG, et al. Mycophenolate mofetil for the treatment of refractory auto-immune haemolytic anaemia and auto-immune thrombocytopenia purpura. Br J Haematol. 2002;117(3):712–715. doi:10.1046/j.1365-2141.2002.03430.x.
- Lin JT, Wang WS, Yen CC, et al. Myelodysplastic syndrome complicated by autoimmune hemolytic anemia: remission of refractory anemia following mycophenolate mofetil. Ann Hematol. 2002;81(12):723–726. doi:10.1007/s00277-002-0539-3.
- Kotb R, Pinganaud C, Trichet C, et al. Efficacy of mycophenolate mofetil in adult refractory auto-immune cytopenias: a single center preliminary study. Eur J Haematol. 2005;75(1):60–64. doi:10.1111/j.1600-0609.2005.00437.x.
- Rao VK, Dugan F, Dale JK, et al. Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome. Br J Haematol. 2005;129(4):534–538. doi:10.1111/j.1365-2141.2005.05496.x.
- O'Connell N, Goodyer M, Gleeson M, et al. Successful treatment with rituximab and mycophenolate mofetil of refractory autoimmune hemolytic anemia post-hematopoietic stem cell transplant for dyskeratosis congenita due to TINF2 mutation. Pediatr Transplant. 2014;18(1):E22–E24. doi:10.1111/petr.12172.
- Jäger U, Barcellini W, Broome CM, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: recommendations from the First International Consensus Meeting. Blood Rev. 2020;41:100648. doi:10.1016/j.blre.2019.100648.
- Besa EC. Rapid transient reversal of anemia and long-term effects of maintenance intravenous immunoglobulin for autoimmune hemolytic anemia in patients with lymphoproliferative disorders. Am J Med. 1988;84(4):691–698. doi:10.1016/0002-9343(88)90106-4.
- Flores G, Cunningham-Rundles C, Newland AC, et al. Efficacy of intravenous immunoglobulin in the treatment of autoimmune hemolytic anemia: results in 73 patients. Am J Hematol. 1993;44(4):237–242. doi:10.1002/ajh.2830440404.