
ABSTRACT
As plants are an essential component of sustainable life support systems, long-duration space missions will require a sophisticated understanding of plant adaptations to spaceflight and microgravity. For many years, transcriptional profiling of steady state mRNA abundances has been used as measure of plant adaptations to the space environment. However, measured changes in transcript abundances are often not reflected in corresponding changes in the proteome due regulatory processes governing translation. Translating ribosome affinity purification (TRAP) is a technique which selectively targets ribosome bound mRNAs for isolation and downstream sequencing. Comparing profiles of ribosome associated mRNAs with total mRNAs provides insight into the translatome and may more accurately inform on the cellular responses to the spaceflight environment. Toward that goal, this work describes a methodology developed ahead of the APEx-07 flight mission.
1. Introduction
Long-distance space travel and colonization is predicated on the cultivation of plants as a source of food, materials, and air purification. Plants however face many challenges to growing and thriving in a spaceflight habitat. In addition to adapting to microgravity, plants must mitigate other hostile environment factors including cosmic radiation, altered gas composition, altered magnetic fields, and varied light levels. Querying the molecular adaptations to these spaceflight related stresses has long been carried out at the transcriptional level by RNA abundance profiling (Paul et al. Citation2017; Manzano et al. Citation2022; Barker et al. Citation2023); first by PCR and microarray based techniques, and now by Next Generation RNA sequencing. However, it is well known that plants, like other eukaryotic systems, employ a complex apparatus of mechanisms to regulate the steps between transcript generation and downstream translation.
The most direct method of characterizing physiologically relevant molecular activities of any organism would ostensibly be to profile entire proteomes in real-time. However, proteomic techniques often require relatively large amounts of tissue, and well-annotated bioinformatic protein data sets. For this reason, relatively few spaceflight plant data sets include a proteomic component (Mazars et al. Citation2014; Ferl et al. Citation2015; Zhang et al. Citation2015; Kruse et al. Citation2020), and fewer still include both a proteomic and transcriptomic component (Kruse et al. Citation2020).
Here we present a methodology for characterizing limited sample masses of spaceflight grown Arabidopsis thaliana with emphasis on the parallel analysis of both Total and Ribosome-Associated RNA species. This methodology aims to simultaneously characterize the transcriptional and translational landscapes of these plants, and by contrasting between the two, provide insight towards the regulatory mechanisms at play.
Translating Ribosome Affinity Purification (TRAP) represents a valuable tool for assessing translatomic abundances (Mazzoni-Putman and Stepanova Citation2018). By this method, endogenous subunits of plant ribosomes are expressed to include an epitope-tagged fusion protein. These subunits are assembled into the ribosomes of plant cells, and may be used by researchers to efficiently isolate active ribosomal complexes, along with their mRNA templates. In Arabidopsis, expression of a FLAG-tagged large ribosomal protein L18 (RPL18) via developmentally regulated promoters has been used to immunopurify ribosome-associated transcripts from specific cell populations of intact plants to characterize floral development (Jiao and Meyerowitz Citation2010) and response to environmental stress (Mustroph et al. Citation2009). A more global proxy of the Arabidopsis translatome is achieved when the His6-FLAG dual epitope tagged RPL18 translational fusion is driven by the cauliflower mosaic virus (CaMV) 35S promoter (Zanetti et al. Citation2005). Powerfully, analysis of ribosome associated transcripts in conjunction with total RNA has been used before to uncover novel translational regulatory mechanisms (Bazin et al. Citation2017).
The objectives of the Advanced Plant Experiment APEx-07 were to characterize the transcriptomic, translatomic, and sRNA landscape of space-grown plants. As a part of this effort, RPL18 was chosen as the sole genotype for flight such that ribosome associated RNA could be isolated and analyzed alongside total and small RNAs.
In this work, we demonstrate that the TRAP-Seq methodology may be adapted to a small scale, suitable for the analysis of limited spaceflight tissue samples. We describe the methods for ribosome immunoprecipitation and RNA recovery which we optimized for our experimental system. Furthermore, we conducted a pilot experiment ahead of the APEx-07 flight mission to compare total and ribosome associated RNA populations isolated from limited quantities of plant root tissue. Protocols were optimized to maximize RNA isolation efficiency and quality, and a Next Generation Illumina Sequencing comparative experiment was carried out.
2. Materials and methods
2.1. RPL18 immunoprecipitation optimization
2.1.1. Immunoprecipitation of RPL18 ribosomal complexes
Transgenic Arabidopsis thaliana (Col-0 background) overexpressing a translational fusion of a ribosomal protein with a His6-FLAG dual epitope tag (RPL18) were obtained from the Arabidopsis Biological Resource Center (Stock No. CS66056).
WT and RPL18 seeds were surface sterilized (see 2.2.1.) and sown on 0.5X MS agar media. Seedlings were grown at 21 °C for a period of 14 days under a 16 h photoperiod (16 h light/8 h dark, 200 µmol m−2 s−1). Whole seedlings were harvested directly into liquid nitrogen and ground to a fine powder. Approximately 250 mg of frozen ground tissue was transferred to pre-chilled glass homogenizers (Pyrex No. 7727-13) and resuspended in 1 mL chilled Polysome Extraction Buffer as described by Zanetti et al. (Citation2005) (200 mM Tris-HCl, pH 8.5, 200 mM KCl, 25 mM EGTA, 35 mM MgCl2, 1% (v/v) Detergent Mix (20% (w/v) Brij-35, 20% (v/v) Triton X-100, 20% (v/v) Nonident P-40, 20% (v/v) Tween-20), 1% (v/v) Polyoxyethylene 10-tridecyl ether, 1 mM DTT, 1% (v/v) Protease Inhibitor Cocktail (Sigma Cat. No. P9599), 50 µg/mL Cycloheximide, 50 µg/mL Chloramphenicol). Tissues were homogenized by grinding in solution for 10 min, on ice. Homogenates were clarified by centrifugation for 10 min at 20,000 RCF at 4 °C and supernatants were transferred to pre-chilled 1.5 mL Eppendorf tubes.
For each isolation, 50 µL Anti-FLAG M2 conjugated magnetic beads (Millipore Sigma Cat. No. M8823) were washed by resuspension in 200 µL Polysome Extraction Buffer. Washed beads were magnetically isolated to the side of the tube by means of a magnetic stand and the buffer was replaced with 50 µL Polysome Extraction Buffer. RPL18 complexes were immunocaptured from clarified homogenates by incubating with washed Anti-FLAG beads at 4 °C, rotating end-over-end, for a period of 2 h. Following incubation, beads were magnetically captured, washed as before in one 200 µL change of Polysome Extraction Buffer, followed by three 200 µL changes of Wash Buffer (200 mM Tris-HCl, pH 8.5, 200 mM KCl, 25 mM EGTA, 35 mM MgCl2, 1 mM DTT, 1% (v/v) Protease Inhibitor Cocktail (Sigma Cat. No. P9599), 50 µg/mL Cycloheximide, 50 µg/mL Chloramphenicol).
For each sample, a 45 µL fraction was reserved from the homogenate at two points in this process – prior to the addition of immunoreactive beads (Input), and after a 2 h incubation with the beads (Unbound). Additionally, 10% of the immunoreactive beads were reserved from the final immunoprecipitation step after washing (Elution).
2.1.2. SDS-PAGE and immunoblot analysis of RPL18 immunoprecipitation fractions
Collected fractions (Input, Unbound, and Elution) were mixed 3:1 with denaturing 4X Laemmli PAGE buffer and incubated in a boiling water-bath for 10 min. Denatured samples were electrophoresed across a 15% Acrylamide SDS-PAGE gel in a Tris/Glycine/SDS buffer system (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3) at 70 V. Proteins were transferred to PVDF membrane in a wet-cell transfer tank using a Tris/Glycine buffer (25 mM Tris, 192 mM glycine, pH 8.3) at 10 V, 4 °C, overnight.
The blot was incubated in 2.5% (w/v) non-fat dry milk in TBST (20 mM Tris pH 7.4, 150 mM NaCl, 0.1% (v/v) Tween-20) for 1 h. FLAG immunoreactive proteins were probed by incubating with horseradish peroxidase conjugated mouse monoclonal α-FLAG M2 antibody (Millipore Sigma Cat. No. A8592) diluted 1:500 in 2.5% (w/v) Milk/TBST for 2 h at RT with gentle shaking. Bands were visualized by incubating in Chemiluminescent substrate (Thermo Fisher Cat. No. 34580) for 5 min and exposing to autoradiography film (Genesee Scientific Cat. No. 30-507). Total protein was visualized post facto by staining the PVDF membrane with India Ink (Levenger PR0220) diluted 1:1000 in TBST for 1 h followed by repeated washing with TBST.
2.1.3. Densitometry based estimation of immunoprecipitation efficacy
A 10 min exposure of autoradiography film (from 2.1.2) was digitalized by a flat-bed scanner in 8-bit grayscale. Input and Unbound bands were analyzed by the Gel Analysis function of FIJI image processing software. Integrated densitometric peak areas were calculated for each and recovery efficiency was defined as (Unbound Peak Area / Input Peak Area) × 100%.
2.2. Experimental Verification Test (EVT) of RPL18 seedlings in VEGGIE hardware
2.2.1. Assembly of petri dishes
Petri dishes were assembled as described by Meyers et al. (Citation2022). Briefly, RPL18 Arabidopsis seeds were surface sterilized by two washes of 70% (v/v) EtOH + Triton X-100 (1 drop Triton X-100 per 100 mL EtOH) followed by a 1 min wash of 95% (v/v) EtOH then allowed to dry. Sterile seeds were adhered to PES membranes in a straight line approximately 5 cm from the top of the plate by 1% aq. (w/v) Guar Gum. The seeded membrane was transferred to 0.5X Murashige and Skoog (MS) agar (0.8% w/v) media, pH 5.7.
2.2.2. Growth parameters for VEGGIE hardware
Assembled petri dishes were stratified in the dark at 4 °C for a period of 5 days. Stratified plates were installed into the VEGGIE hardware for germination and growth. A 16 h photoperiod (16 h light/8 h dark) was maintained at an intensity of 200 µmol m−2 s−1 for 12 days. Throughout the germination and growth of the seedlings, an ambient temperature of 21 °C was maintained.
2.2.3. Tissue harvest and stowage
Following growth, petri dishes were disassembled from the VEGGIE unit and PES membranes of grown seedlings were transferred to the petri dish lids and wrapped in aluminum foil. Harvested samples were temporarily held in cold stowage bags (−130 °C) and then transferred to an ultra-low cryogenic freezer (−80 °C). Frozen samples were later shipped from Kennedy Space Center to Raleigh, NC on dry ice (−78.5 °C) and transferred to another cryogenic freezer (−80 °C) until dissection.
2.3. Isolation of total and ribosome associated RNA species
2.3.1. Tissue dissection and initial homogenization
Frozen membranes of RPL18 seedlings were removed from cryogenic stowage and transferred to sit directly atop a pre-chilled (−80 °C) block of aluminum. Shoot and root fractions were quickly dissected from each membrane by fracturing each seedling at the hypocotyl junction with a scalpel while still frozen. Each fraction was transferred to liquid nitrogen and ground to a fine powder using a mortar and pestle.
2.3.2. Isolation of total RNA
Total RNA was isolated from three replicate fractions of ground root tissues (one EVT petri plate each, ∼30 mg tissue each) using a silica column based kit (Norgen Cat. No. 25800) according to the manufacturer’s directions.
2.3.3. Isolation of ribosome associated RNA
Ribosome associated RNA was isolated from three replicate fractions of ground root tissues (one EVT petri plate each) by the method described in 2.1.1. RPL18 immunocomplexes, and their associated mRNAs, were released from the beads by resuspending in 650 µL Lysis Buffer (Norgen Cat. No. 25800) supplemented with 10 µL/mL β-Mercaptoethanol and incubating at 4 °C, rotating end-over-end for 10 min. Magnetic beads were removed from the solution and ribosome associated mRNAs were subsequently purified from the Lysis Buffer by a silica column based kit (Norgen Cat. No. 25800), according to the manufacturer’s directions and eluted in 50 µL molecular grade H2O.
2.3.4. RNA sequencing
Three replicate isolates each of total and polysomal RNA were prepared from the same EVT experiment. RNA samples were submitted to the North Carolina State Genomic Sciences Laboratory (GSL) (Raleigh, NC, USA) for Illumina RNA library construction and sequencing which was carried out by the GSL standard protocols as described in Luis et al. (Citation2022), with modifications. Briefly, RNA integrity, purity, and concentration were assessed using an Agilent 2100 Bioanalyzer with an RNA 6000 Nano Chip (Agilent Technologies, USA). Purification of messenger RNA (mRNA) was performed using the oligo-dT beads provided in the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs, USA). Complementary DNA (cDNA) libraries for Illumina sequencing were constructed using the NEBNext Ultra Directional RNA Library Prep Kit (NEB) and NEBNext Multiplex Oligos for Illumina (NEB) using the manufacturer-specified protocol. The amplified library fragments were purified and checked for quality and final concentration using an Agilent 2200 Tapestation (Agilent Technologies, USA). The final quantified libraries were pooled in equimolar amounts for clustering and sequencing on an Illumina NovaSeq 6000 DNA sequencer, utilizing a 150 × 2 bp paired end sequencing reagent kit (Illumina, USA). The software package Real Time Analysis was used to generate raw bcl files, which were then de-multiplexed by sample into fastq files for data submission.
2.3.5. Transcriptomic analysis
Primers and low-quality sequences were removed from reads using Trim Galore! 0.6 under the base parameters. Paired reads were aligned using version 2.6 of STAR and the Araport11 genome annotations (Cheng et al. Citation2017). Gene counts were then normalized using the trimmed mean of M values (TMM) as calculated using limma v3.52 voom. Unmodified Gene counts and normalized gene were both used for the analyses of libraries. Genes with extremely low expression (less than 15 transcripts across all three biological replicate) were excluded from the analyses to avoid noise potentially introduced by inaccurate alignment or extreme ratios that represent a miniscule fraction of the total library. Approximate centromere location was determined from the initial Arabidopsis genome construction (The Arabidopsis Genome Initiative Citation2000).
3. Results
3.1. RPL18 immunoprecipitation optimization
Given the limited tissue mass of spaceflight samples, it was essential that we optimize a protocol for the efficient recovery of ribosome associated mRNA species. We developed an immunoprecipitation method which prioritizes efficient sample handling with minimal loss due to washing etc. To accomplish this, we chose to employ a paramagnetic resin covalently conjugated directly to an α-FLAG antibody. Commercially prepared stocks of this resin were able to bypass the need to prepare fresh immunoreactive resins from monoclonal antibodies and protein G before every isolation, and were consistent from batch to batch. The ability of these resins to be magnetically sequestered into a tight pellet without centrifugation allowed rapid and efficient washing steps, with minimal loss compared to traditional agarose resins.
Immunoblot analysis of collected fractions indicate that RPL18 ribosomal complexes are efficiently immunoprecipitated by this method. A ∼23.5 kDa sized band was detected specifically in RPL18 seedling homogenates, and not in WT controls (). Immunoprecipitation of this species using the same antibody, conjugated to a super paramagnetic resin sequestered the soluble protein complex efficiently in a relatively short period of time (2 h). Densitometric analysis compared the abundance of this species after incubation (Unbound) with free levels prior to immunoprecipitation (Input) and estimates a recovery efficiency of 94%.
Figure 1. Immunoblot of Input, Unbound, and Elution fractions of the RPL18 immunoprecipitation protocol.
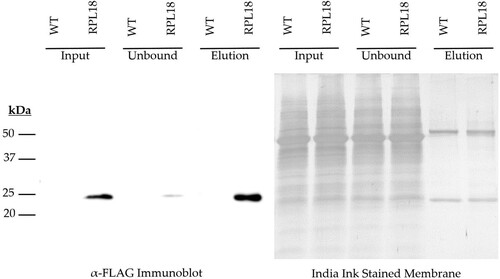
Interestingly, this species was tightly bound by the immunoreactive resin, and not liberated by standard acidic elution methods (Glycine pH 2.6, Appendix A Figure 1). Incubation with the denaturing reagent β-mercaptoethanol, coupled with boiling however was found to efficiently liberate the immunocaptured species, and was detectable by immunoblot. It was further found that this dissociation could also be achieved by incubating washed immunoreactive resins directly in a chaotropic lysis buffer supplemented with β-mercaptoethanol as the first step of silica column based nucleic acid isolation protocols.
3.2. Ribosome associated and total RNA isolation from VEGGIE grown pre-flight test materials
To verify the suitability of this TRAP-seq methodology for use in spaceflight grown Arabidopsis experiments, we conducted a small scale experiment sourcing frozen plant material from plates grown in the VEGGIE apparatus as part of the APEx-07 Experiment Verification Test (EVT). Three frozen plates of plant material were dissected into shoot and root fractions and masses were recorded ().
Table 1. Frozen tissue weights of starting materials from Experiment Verification Test.
Frozen tissues were immediately processed by the immunoprecipitation method describe above, and recovered nucleic acid fractions were analyzed for quality and quantity by BioAnalyzer microfluidic capillary electrophoresis (). All processed samples exhibited a high degree of mass recovery and quality, and were deemed suitable for downstream analysis by Next Generation Illumina Sequencing. In parallel, an additional set of three biological replicate plate roots produced total RNA of similar quality ().
Table 2. Recovered ribosome associated RNA mass quantity and RNA Integrity (RIN) values.
Table 3. Recovered total RNA mass quantity and RNA Integrity (RIN) values.
3.3. Next generation RNA sequencing
APEx-07 is intended to analyze both total RNA and ribosome associated RNAs in parallel from the same experiment. From these analyses, the experiment aims to examine and contrast potential differences between the transcriptional and translational landscapes of spaceflight grown plants.
To validate our experimental pipeline prior to the flight experiment, we conducted a small scale RNA sequencing experiment with our most limiting tissue type – roots. In addition to ribosome associated RNA, total RNA was isolated directly from roots of the same EVT experiment using the same silica column based isolation kit for both purifications. In parallel, three bioreplicate isolates of total and ribosome associated RNA populations were prepared for Illumina sequencing.
3.4. Sequence analysis and findings
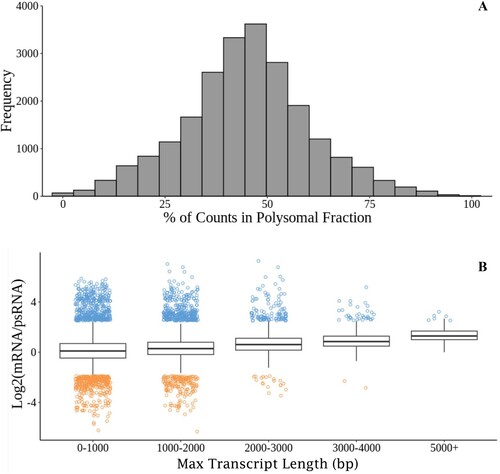
Both the total mRNA and polysomal extractions produced libraries of aligned reads of equivalent sizes (Welch two sample t-test, p = 0.927). Of the 25,102 unique loci returned after alignment, 93% of genes detected had transcripts present in both fractions. The relative proportion of transcripts followed a normal distribution ((A)). Because transcripts with multiple ribosomes attached could have a higher chance of being pulled out during immunoprecipitation, there was a chance that longer transcripts with more area for ribosomes to attach might be enriched in the polysomal fraction. There was not an appreciable increase in the number of long reads found in the polysomal fraction. Instead, transcripts followed the same trend of most reads being more abundant in the total RNA fraction, even in the potentially longest transcripts ((B)).
Figure 2. Relationships between polysomal and mRNA fractions. A. Frequency of genes expressed in the polysomal fraction divided by all mRNA aligned (total mRNA plus polysomal mRNA) for that gene. B. Log2 of the ratio of the total mRNA divided by the polysomal mRNA across the maximum potential lengths of transcripts. Ratios outside of the inter-quartile ranges are plotted in blue (mRNA higher) or orange (psRNA higher) to visualize the variance in data.
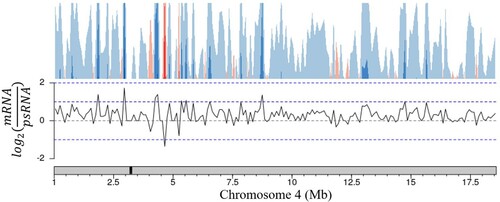
These trends in expression are also conserved across nuclear chromosomes. The few areas with higher polysomal representation are randomly spread across chromosomes with no apparent bias towards chromosomal region (). Taken together these results show that both the total mRNA and polysomal mRNA fractions produce high quality read libraries with equivalent ability to detect transcripts regardless of transcript length or the location of the encoding loci.
Figure 3. Ratio of gene representation in either the mRNA fraction or polysomal RNA fraction across a representative chromosome. Chromosomes were tiled into 100,000 bp regions, average ratios were taken for each tile. The approximate centromere location is marked in black on the chromosome.
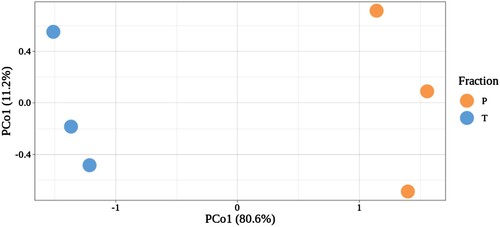
Even with the consistencies in libraries produced from the same tissue under the same growth conditions, the two methods produced distinct populations of transcripts (). Both the total mRNA and the polysomal fractions produced high quality libraries with no bias in the nature of the reads returned. Each library represent a unique population of RNA showing that the polysomal and mRNA extractions were successful in representing unique biological states.
Figure 4. Principal coordinate analysis (PCoA) of three biological replicates of total (T) and ribosome associated (P) reads.
4. Discussion
Advances in sequencing technologies, computing power, and bioinformatics have welcomed in a new era of powerful and relatively cost-effective research practices. As platforms improve, sequencing costs, and required amounts of starting material have fallen dramatically. However, it is well understood that the central dogma of eukaryotic cells is regulated at multiple levels. Although traditional transcriptomic analysis of spaceflight grown plants represents a great tool in our exploration of their unique adaptations, it leaves out some of those regulatory underpinnings which govern function.
While the transcription of various mRNAs is a ubiquitous and on-going activity in any eukaryotic cell, translation of these transcripts to functional proteins is concomitant with specific recruitment to polysomal complexes. This highly regulated process is often complex and difficult to study. Coupling TRAP-Seq and RNA-Seq strategies together allows for a more complete understanding of the translational/translatomic interface. Thus, a combined approach may illuminate some of the regulatory mechanisms at play.
Often, spaceflight grown plant tissues are of limited quantities owing to the nature of hardware and transport constraints associated with spaceflight research. As such, transcriptional sequencing based methods as opposed to mass-spec based proteomics still offer one of the most tissue efficient means of extracting useable data from these experiments.
Our efforts, here described, aim to make the most of limited tissue quantities. As such, particular care was made to improve upon the existing TRAP-Seq protocols published thus far. One of the seemingly wasteful decisions described in our methodology can be found in the direct transfer of washed immuno-resins to a denaturing chaotropic solution upstream of nucleic acid purification. This elution strategy effectively destroys the immuno-resin, separating the heavy and light chains of the mAb used for immunoprecipitation. However, we have found this to be worth the costs; this step minimized the handling time between initial immunocapture and downstream nucleic acid purification, and maximized the yields recovered therein. By adopting this strategy we were able to reliably isolate high quality RNA, unadulterated by degradation events associated with whole-tissue homogenates.
As a means of assessing the transcriptomic and translatomic landscapes of plant adaptations to spaceflight, parallel RNA-seq and TRAP-seq as described in this methodology offer powerful insight. While this Experimental Verification Test did not include a microgravity or gravistimulation treatment, data from this experiment indicate that transcriptional and translatomic abundances may be reasonably assessed in this manner. From a limited amount of tissue, these procedures can simultaneously generate two unique, robust datasets. Moreover, in the case of a spaceflight experiment, comparison of these datasets may provide understanding of more complex translational regulatory mechanisms underpinning plant adaptations to the spaceflight environment.
Acknowledgements
The authors would like to thank the crew at Kennedy Space Center, especially Gerard Newsham, Susan Manning-Roach, Anne Marie Campbell, and Lucy Orozco for their support with preflight preparation and ground operations.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Barker R, Kruse CPS, Johnson C, Saravia-Butler A, Fogle H, Chang H-S, Trane RM, Kinscherf N, Villacampa A, Manzano A, et al. 2023. Meta-analysis of the space flight and microgravity response of the Arabidopsis plant transcriptome. NPJ Micrograv. 21. doi:10.1038/s41526-023-00247-6.
- Bazin J, Baerenfaller K, Gosai SJ, Gregory BD, Crespi M, Bailey-Serres J. 2017. Global analysis of ribosome-associated noncoding RNAs unveils new modes of translational regulation. Proc Natl Acad Sci USA. 114:E10018–E10027. doi:10.1073/pnas.1708433114.
- Cheng CY, Krishnakumar V, Chan AP, Thibaud-Nissen F, Schobel S, Town CD. 2017. Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 89:789–804. doi:10.1111/tpj.13415.
- Ferl RJ, Koh J, Denison F, Paul AL. 2015. Spaceflight induces specific alterations in the proteomes of arabidopsis. Astrobiology. 15:32–56. doi:10.1089/ast.2014.1210.
- Jiao Y, Meyerowitz EM. 2010. Cell-type specific analysis of translating RNAs in developing flowers reveals new levels of control. Mol Syst Biol. 6:1–14. doi:10.1038/msb.2010.76.
- Kruse CPS, Meyers AD, Basu P, Hutchinson S, Luesse DR, Wyatt SE. 2020. Spaceflight induces novel regulatory responses in Arabidopsis seedling as revealed by combined proteomic and transcriptomic analyses. BMC Plant Biol. 20:237. doi:10.1186/s12870-020-02392-6.
- Luis JM, Carbone I, Mack BM, Lebar MD, Cary JW, Gilbert MK, Bhatnagar D, Wientjes CC, Payne GA, Moore GG, et al. 2022. Dataset for transcriptomic profiles associated with development of sexual structures in Aspergillus flavus. Data Brief. 42:108033. doi:10.1016/j.dib.2022.108033.
- Manzano A, Carnero-Diaz E, Herranz R, Medina FJ. 2022. Recent transcriptomic studies to elucidate the plant adaptive response to spaceflight and to simulated space environments. iScience. 25. doi:10.1016/j.isci.2022.104687.
- Mazars C, Brière C, Grat S, Pichereaux C, Rossignol M, Pereda-Loth V, Eche B, Boucheron-Dubuisson E, Le Disquet I, Medina FJ, et al. 2014. Microsome-associated proteome modifications of Arabidopsis seedlings grown on board the international space station reveal the possible effect on plants of space stresses other than microgravity. Plant Signal Behav. 9. doi:10.4161/psb.29637.
- Mazzoni-Putman SM, Stepanova AN. 2018. A plant biologist’s toolbox to study translation. Front Plant Sci. 9:1–20. doi:10.3389/fpls.2018.00001.
- Meyers A, Land E, Perera I, Canaday E, Wyatt SE. 2022. Polyethersulfone (PES) Membrane on Agar Plates as a Plant Growth Platform for Spaceflight. Gravit Space Res. 10:30–36. doi:10.2478/gsr-2022-0004.
- Mustroph A, Zanetti ME, Jang CJH, Holtan HE, Repetti PP, Galbraith DW, Girke T, Bailey-Serres J. 2009. Profiling translatomes of discrete cell populations resolves altered cellular priorities during hypoxia in Arabidopsis. Proc Natl Acad Sci USA. 106:18843–18848. doi:10.1073/pnas.0906131106.
- Paul AL, Sng NJ, Zupanska AK, Krishnamurthy A, Schultz ER, Ferl RJ. 2017. Genetic dissection of the Arabidopsis spaceflight transcriptome: are some responses dispensable for the physiological adaptation of plants to spaceflight? PLoS One. 12:1–24. doi:10.1371/journal.pone.0180186.
- The Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 408:796–815. doi:https://doi.org/10.1038/35048692.
- Zanetti ME, Chang I-F, Gong F, Galbraith DW, Bailey-Serres J. 2005. Immunopurification of polyribosomal complexes of arabidopsis for global analysis of gene expression. Plant Physiol. 138:624–635. doi:10.1104/pp.105.059477.
- Zhang Y, Wang L, Xie J, Zheng H. 2015. Differential protein expression profiling of Arabidopsis thaliana callus under microgravity on board the Chinese SZ-8 spacecraft. Planta. 241:475–488. doi:10.1007/s00425-014-2196-x.
Appendices
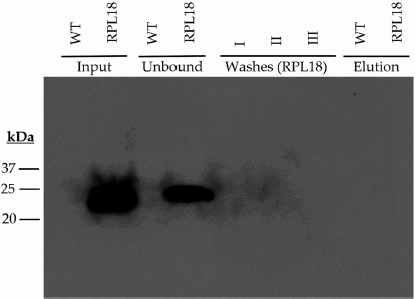
Appendix A.α-FLAG immunoblot showing failure of RPL18 complex to elute by standard Glycine pH 2.6 acidic elution methods.
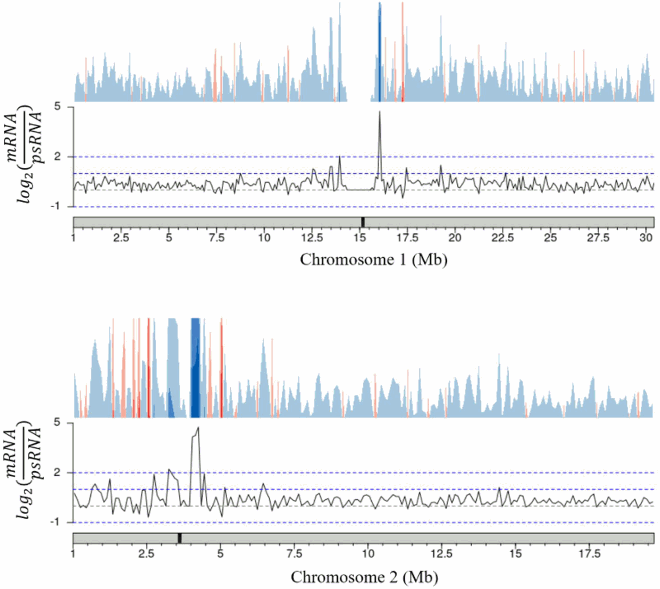
Appendix B. Total mRNA / polysomal mRNA ratio analysis, chromosomes 1, 2, 3, 5. (Chromosome 4 shown in main text).
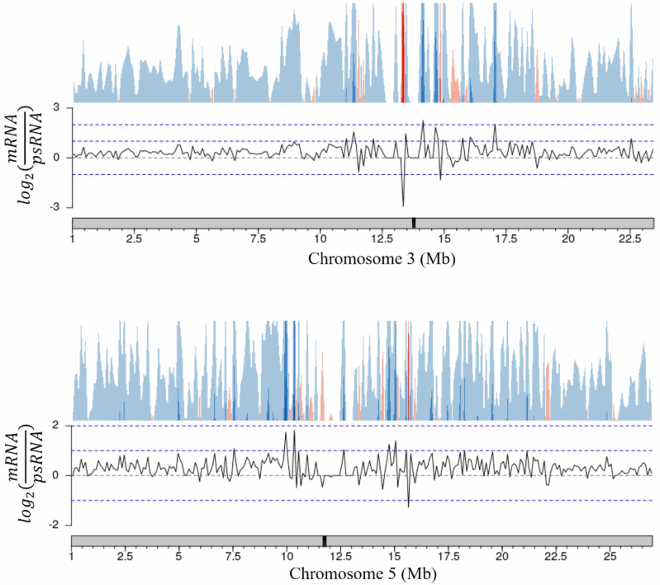
Appendix B. Continued.

Appendix C. Representative image of 12 d old plant material grown in VEGGIE plate system prior to cryogenic stowage and downstream harvest and processing.