
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Introduction
The field of RNA-targeted small molecules is rapidly evolving, owing to the advances in experimental and computational technologies. With the identification of several bioactive small molecules that target RNA, including the FDA-approved risdiplam, the biopharmaceutical industry is gaining confidence in the field. This review, based on the literature obtained from PubMed, aims to disseminate information about the various technologies developed for targeting RNA with small molecules and propose areas for improvement to develop drugs more efficiently, particularly those linked to diseases with unmet medical needs.
Areas covered
The technologies for the identification of RNA targets, screening of chemical libraries against RNA, assessing the bioactivity and target engagement of the hit compounds, structure determination, and hit-to-lead optimization are reviewed. Along with the description of the technologies, their strengths, limitations, and examples of how they can impact drug discovery are provided.
Expert opinion
Many existing technologies employed for protein targets have been repurposed for use in the discovery of RNA-targeted small molecules. In addition, technologies tailored for RNA targets have been developed. Nevertheless, more improvements are necessary, such as artificial intelligence to dissect important RNA structures and RNA–small-molecule interactions and more powerful chemical probing and structure prediction techniques.
1. Introduction
Historically, drug discovery programs have focused on developing small molecules that target proteins to alter the outcome of biological processes. However, according to a recent survey, less than 1% of the total number of human proteins remain as viable new targets for small-molecule drugs [Citation1]. The survey noted that a large portion of disease-related proteins (roughly 60%) are considered ‘undruggable’ because they lack distinctive clefts or pockets capable of binding small-molecule drugs. Given this diminishing pool of druggable proteins, many are looking into the possibility of targeting RNA with small molecules.
Figure 1. Why target RNA with small molecules? (a) RNAs perform a myriad of functions, which are often mediated by structurally conserved motifs. The HIV-1 transactivation response (TAR), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) stem loop 5 (SL5), and the flavin mononucleotide (FMN) riboswitch, whose secondary structures are shown, are a few examples of functional RNA motifs that are structurally conserved. (b) Many bioactive small molecules that act on RNA targets have now been identified. The equilibrium dissociation constant (Kd), 1.5 maximal effective concentration (EC1.5) or median inhibitory concentration (IC50) are provided. (c) Like the proteome, the transcriptome is likely to contain many druggable sites [Citation2], as the ones depicted in red spheres. The druggable sites on the FMN riboswitch (PDB ID: 3 f4g) [Citation3], add adenine riboswitch (PDB ID: 5swe) [Citation4], simian retrovirus type-1 (SRV-1) pseudoknot (PDB ID: 1e95) [Citation5], enterovirus 71 internal ribosomal entry sites (IRES) (PDB ID: 6xb7) [Citation6], HIV-1 TAR (PDB ID: 1qd3) [Citation7], UGGAA/UGGAA pentad in spinocerebellar ataxia type 31 (SCA31) [Citation8] were calculated using Fpocket [Citation9].
![Figure 1. Why target RNA with small molecules? (a) RNAs perform a myriad of functions, which are often mediated by structurally conserved motifs. The HIV-1 transactivation response (TAR), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) stem loop 5 (SL5), and the flavin mononucleotide (FMN) riboswitch, whose secondary structures are shown, are a few examples of functional RNA motifs that are structurally conserved. (b) Many bioactive small molecules that act on RNA targets have now been identified. The equilibrium dissociation constant (Kd), 1.5 maximal effective concentration (EC1.5) or median inhibitory concentration (IC50) are provided. (c) Like the proteome, the transcriptome is likely to contain many druggable sites [Citation2], as the ones depicted in red spheres. The druggable sites on the FMN riboswitch (PDB ID: 3 f4g) [Citation3], add adenine riboswitch (PDB ID: 5swe) [Citation4], simian retrovirus type-1 (SRV-1) pseudoknot (PDB ID: 1e95) [Citation5], enterovirus 71 internal ribosomal entry sites (IRES) (PDB ID: 6xb7) [Citation6], HIV-1 TAR (PDB ID: 1qd3) [Citation7], UGGAA/UGGAA pentad in spinocerebellar ataxia type 31 (SCA31) [Citation8] were calculated using Fpocket [Citation9].](/cms/asset/c7b3b3bf-2d2b-40cc-80b2-dddb4028fd36/iedc_a_2134852_f0001_oc.jpg)
RNA molecules were previously viewed as mere messengers between DNA and protein synthesis. Today, we know that RNA has catalytic and regulatory functions in diverse biological events ()). As such, RNA presents boundless opportunities for intervention with small molecules. For example, small molecules that target mRNAs could control the production of disease-related proteins, even those that were heretofore deemed undruggable. In addition, the roles of non-coding RNAs in disease are gradually being substantiated [Citation10]. Since non-coding RNAs occupy a significant fraction of the human genome [Citation11], targeting them would broaden the reach of small-molecule drugs.
The concept of targeting RNAs with small molecules is not a novel one. In the late 1980s, it was discovered that the target of aminoglycoside antibiotics are bacterial ribosomal RNAs [Citation12]. Thereafter, aminoglycosides were revealed to promiscuously bind RNA [Citation13,Citation14], and while some viewed this promiscuity as an opportunity to optimize aminoglycosides for targeting other RNAs, many opted to divert to more drug-like small molecules. Thus far, more than 200 bioactive small molecules were reported to bind RNA molecules with diverse structural motifs ()) [Citation15,Citation16]. The RNA binders can be classified into two: (1) ‘traditional’ small molecules with properties satisfying Lipinski’s rules (MW ~ 500 Da) and (2) larger, multivalent ligands (~2000 Da). The consensus is that drug-like small molecules would bind structurally complex RNAs with protein-like binding pockets, while multivalent ligands would bind low-complexity RNAs ()) [Citation17,Citation18].
This review will focus on the early stages of RNA-targeted small-molecule drug discovery and describe the experimental and computational tools that can be used in each stage. First, the strategies for identifying potentially functional and druggable RNA motifs will be delineated. Next, several high-throughput screening (HTS) approaches and biophysical techniques for hit validation will be highlighted, and the factors to consider for each of them will be discussed. Then, the methods for assessing the functional effects and target engagement of RNA-binding small molecules will be described. Finally, an overview of the techniques for determining the three-dimensional (3D) structures of RNA–small-molecule complexes, along with ways by which such structural information can be exploited to accelerate the drug discovery process, will be given. The references for this review were obtained through searches of the PubMed database (www.pubmed.gov) through July 2022.
2. Identification of RNA targets
Most efforts to discover RNA-targeted small molecules have been directed against already-known functional RNA motifs, such as HIV-1 transactivation response (TAR) [Citation19], the flavin mononucleotide (FMN) riboswitch [Citation20–22], the severe acute respiratory syndrome coronavirus frameshifting pseudoknot [Citation23], viral internal ribosomal entry sites (IRES) [Citation8,Citation24], and disease-associated RNA repeat sequences [Citation25,Citation26]. However, there remains a plethora of therapeutically valuable RNA motifs in the transcriptome waiting to be unveiled. The identification of target motifs on RNA molecules can be approached using the following strategy: (1) extraction of motifs on an RNA molecule, (2) analysis of RNA secondary structures, and (3) assessment of RNA motifs to select the most suitable target(s).
2.1. Extraction of RNA motifs
Motifs on an RNA can be extracted using secondary structure prediction () and chemical-probing approaches. The mainstream method in RNA secondary structure prediction uses dynamic programming to calculate an optimal secondary structure with the minimum free energy (MFE) among all possible outcomes [Citation39,Citation57]. The free energy is typically estimated from the thermodynamic free energies for the base-pairing stems and the conformational entropies for the unpaired loop regions [Citation58,Citation59]. The accuracy of secondary structure predictions using free energy minimization can vary based on the length of the RNA; for RNA sequences shorter than 700 nucleotides, the prediction accuracy is approximately 70%, but for longer RNAs, the accuracy decreases [Citation60].
Table 1. List of programs for RNA secondary structure prediction.
Another common method for RNA secondary structure prediction is comparative sequence analysis. It is generally believed that RNA motifs whose biological functions are dictated by structure show high structural conservation when compared to the same RNA of related species [Citation61]. Thus, in comparative sequence analysis, base-pairing stems and loops are identified based on their occurrence in identical or similar locations on homologous RNAs. While comparative sequence analysis is exceptionally accurate at predicting secondary structures (>95% accuracy), it may not be applicable to all RNAs and requires careful multiple sequence alignments [Citation62].
The most widely used chemical probing methods employ dimethyl sulfate (DMS), which modifies the bases of non-Watson–Crick paired or single-stranded nucleotides [Citation63], and selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE) reagents, which modifies the ribose groups of dynamic nucleotides [Citation64,Citation65]. The results of DMS and SHAPE chemical probing are read out using primer extension-truncation and adapter-ligation to create libraries to be analyzed by massive parallel sequencing. However, the reactivities obtained from these methods may be inaccurate due to: (1) low signal levels, (2) biases introduced by the truncation–adapter-ligation steps [Citation66,Citation67], (3) the presence of naturally occurring modified nucleotides and stable secondary structures that result in unwanted truncation, and (4) improper statistical analysis. Thus, DMS mutational profiling with sequencing (DMS-MaPseq) [Citation68] and SHAPE-MaP ()) were developed to overcome these limitations.
Figure 2. Methods for identification of RNA targets. (a) SHAPE-MaP can be used to probe RNA secondary structures on a massively parallel scale. (b) MobyDick enables the analysis of RNA secondary structure predictions. When the analysis is completed, the MobyDick interface displays the locations, secondary structure, global population (PGlobal), and stability (∆G) of the motifs.
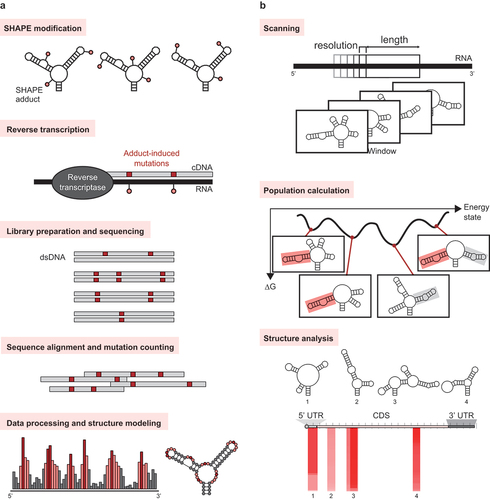
In the MaP strategies, the modified nucleotides are detected by mutational profiling. MaP takes advantage of conditions where reverse transcriptases misread modified nucleotides and incorporate non-complementary nucleotides into the original sequence in the newly generated cDNA. These cDNAs are amplified using methods that are easier to execute and produce less bias compared to truncation–adapter-ligation strategies [Citation69,Citation70]. Then, the amplified cDNA sequences are subjected to massively parallel sequencing, and the MaP data are analyzed [Citation71] to produce a reactivity profile.
The chemical reactivities from any of the above probing experiments can be incorporated as constraints in several RNA secondary structure prediction software, such as RNAstructure [Citation46,Citation47] and RNAfold in the Vienna RNA package [Citation42,Citation43]. Mathews et al. reported an average 9% increase in accuracy (from 67% to 76%) of secondary structure prediction with the incorporation of chemical reactivity constraints [Citation34].
2.2. Analysis of RNA secondary structures
After extracting the RNA motifs, the next step would be analyzing the RNA secondary structures to narrow down the list of candidate target RNA motifs. We created a software, MobyDick, which is a part of our ibVIS platform technology, for analyzing the results of secondary structure predictions from any software using a scanning window ()) [Citation72]. First, the results of the secondary structure prediction are input into MobyDick, and the set of predicted secondary structures for each user-defined window, moved at a user-defined resolution from the 5’- to the 3’-end of the RNA sequence, are gathered. Next, the population of each motif found within the predicted structures for every window (local population or ) is calculated using Maxwell-Boltzmann statistics. Finally, the motif’s global population (
) value, which indicates the probability that the motif exists, is generated using the following equation:
where is the weight function for the window
. Assuming that the ribosomal speed along the mRNA is constant,
is equal to
. To date, we have used MobyDick for the analysis of roughly 100 transcripts, and motifs with
values greater than 50% showed high agreement with experimentally derived secondary structures.
The Moss group recently developed ScanFold [Citation73], another RNA secondary structure analytic tool that uses the scanning window approach. ScanFold gives a z-score, which compares the MFE of a sequence within an RNA to the average MFE of a set of randomized RNA sequences normalized by the standard deviation of the MFE. Motifs with more negative z-scores (< −2) are considered very stable and likely to perform a biological function. ScanFold and MobyDick differ not only in terms of the kind of output they provide (i.e. z-score versus PGlobal) but also in that the former utilizes only RNAFold while the latter can accept the results any secondary structure prediction software. This confers MobyDick an advantage in that the use of several software in parallel can compensate for the computational bias of each software and increase one’s confidence in the motifs predicted should they be identified by multiple software.
2.3. Target RNA motif selection
Once the number of candidate target RNA motifs is pared down to the most promising, one can assess individual motifs more thoroughly. The location of the motif on an mRNA or pre-mRNA provides clues to its function. In general, stable motifs on the 5’ untranslated region (UTR) may regulate translation [Citation74–79], those on the junctions between the exons and introns may be involved splicing [Citation80–82], and those on the 3’ UTR may control degradation and stabilization processes [Citation83–86]. From a survey of the complete genomes of 340 species, Gu et al. [Citation87] posited that stable RNA motifs near the start codon regulate translation by interfering with start codon recognition. It was also reported that highly stable motifs on the coding region may stall the ribosome and activate the ribosome-associated protein pathway, which targets newly synthesized proteins for proteasomal degradation [Citation88]. As a rule of thumb, we select stable RNA motifs on the 5’ UTR and the coding region of the mRNA and splicing relevant RNA motifs on exon-intron junctions of the pre-mRNA as targets of small-molecule drug discovery. It is also worth considering motifs on the 3’ UTR and non-coding RNAs after verifying their therapeutic implications with experimental and computational approaches. In addition, we select RNA motifs that bind proteins, which are components of cellular metabolism or regulatory networks, as suggested by Warner et al. [Citation17].
3. High-throughput screening of compounds that bind to RNA motifs
Several HTS approaches have been used to identify RNA-binding small molecules, and they can be characterized as target-based or phenotypic (, ). Target-based screening approaches usually employ biophysical techniques with the goal of identifying small molecules that bind to a known target. In contrast, in phenotypic screening, the target is unknown, and the goal is to screen for compounds that alters the phenotype of a cell or organism.
Figure 3. High-throughput screening approaches for identifying small molecule compounds that bind to RNA. (a) In fluorescence-based screening, a fluorescently labeled molecule – either the RNA target or an indicator – is used. Fluorescence resonance energy transfer (FRET) employs an RNA probe that is labeled with a fluorophore and a quencher. The FRET RNA probe is combined with the screening compounds and subjected to heating. The melting temperatures (Tm) of the RNA are measured using qPCR. Compounds that increase the Tm of the RNA bind to and stabilize the RNA. (b) Affinity-based screening methods do not require labeling of the RNA target. The automated ligand identification system (ALIS) is an indirect affinity-based screening method that involves the equilibration of the target RNA with a mixture of the screening compounds, size exclusion chromatography to purify the RNA–small molecule complexes, reverse-phase HPLC to dissociate the small molecules from the RNA, and mass spectrometry to identify the small molecule hits. (c) In small molecule microarray screening, the small molecules are immobilized onto microarray plates and used for screening against RNA targets. The hits are analyzed, e.g. by detecting increase in fluorescence if the RNA target is fluorescently labeled. (d) Fragment-based screening employs biophysical techniques, usually NMR, to identify low molecular weight compounds that bind to RNA targets. In 19F-NMR-based fragment screening, the 1D NMR spectra of mixtures of fluorinated compounds, before and after the addition of the RNA target, are taken. The hits are identified by looking for changes in the NMR signals of the fluorinated compounds; RNA-binding compounds exhibit more pronounced NMR signals after the addition of RNA. (e) In a typical DNA-encoded library (DEL) screening against RNA targets, the RNA is immobilized and incubated with small molecule compounds conjugated to a DNA barcode. The DEL members that bind to the RNA are eluted, and their identities are revealed after DNA amplification and sequencing. (f) Docking is the most widely used virtual screening approach. It requires that the three-dimensional structure of the RNA target be known so that the binding energies between the RNA and the screening compounds can be predicted in silico. (g) Most of the well-known RNA-targeted small molecules have been identified using phenotypic screening. In this method, the small molecules are evaluated for their ability to produce the desired phenotype within a cellular environment. The identity of RNA target is only revealed after molecular target identification and mechanistic studies.
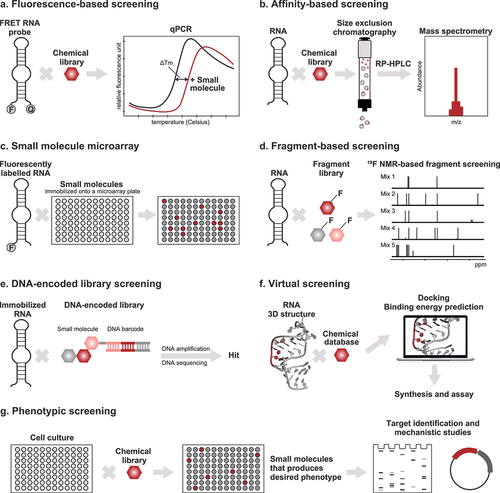
Table 2. Selected screens conducted against RNA targets*.
Some measures that are prerequisite to successful HTS campaigns include: (1) the rigorous design and development of the assay, (2) the use of positive and negative controls for checking the quality of the assay, and (3) the use of compound libraries with wide chemical diversity. In addition, the merits of a properly designed and executed HTS assay would inevitably be wasted if appropriate statistical methods for hit selection are not utilized. Furthermore, the statistical methods for analysis of HTS data must be adapted to the characteristics of the HTS approach and to the responses observed.
Regardless of whether target-based or phenotypic screening approaches are used, it is advisable to perform a cascade of experiments to select the specific and high-quality hits from the HTS active compounds. Since HTS is typically done at a single compound concentration, subsequent dose-dependence experiments using a broad concentration range are ideal. The dose–response curves would not only give the IC50 values but also provide information about the compound’s toxicity and solubility. In addition, counter screens can be used to assess the specificity of the HTS active compounds.
3.1. Fluorescence-based screening
The three fluorescence-based assays that are often employed for screening of RNA binders are: (1) fluorescence resonance energy transfer (FRET)-based assay ()), (2) time-resolved FRET (TR-FRET) assay, and fluorescent indicator displacement (FID). FRET refers to the transfer of energy from a donor fluorophore to an acceptor fluorophore conjugated to the target biomolecule. FRET-based assays are convenient and extremely sensitive and have become popular for screening small-molecule libraries against RNA targets. Simone et al. [Citation92] used a FRET-based assay to screen for small-molecule stabilizers of the C9orf72 (G4C2)4 G-quadruplex RNA, which is a known cause of frontotemporal dementia and amyotrophic lateral sclerosis [Citation116]. The authors monitored the changes in melting temperature of the 5’-FAM and 3’-TAMRA labeled (G4C2)4 RNA upon heating in the presence of small molecules and identified three structurally similar small molecules that stabilize the RNA. The small molecules were subsequently shown to reduce the frequency of RNA foci and the levels of dipeptide repeat protein in C9orf72 patient neurons. Furthermore, the most effective small molecule, DB1273, was found to improve survival and reduce levels of toxic poly-(glycine-arginine) in C9orf72 flies.
The TR-FRET assay combines the low background aspect of time-resolved fluorimetry with the homogenous format of FRET. Thus, TR-FRET provides increased flexibility, reliability, sensitivity, and throughput. TR-FRET was used to identify small molecules that inhibit the formation of the complex between (CUG)n expansion repeat RNAs with muscleblind-like splicing regulator 1 (MBNL1) [Citation93,Citation117,Citation118] that causes myotonic dystrophy type 1 (DM1). After combining biotinylated (CUG)12 RNA and MBNL1-His6 protein, fluorescently labeled streptavidin and anti-His6 antibody were added to the solution. Streptavidin binds to the biotinylated (CUG)12 RNA whereas the anti-His6 antibody binds to MBNL1. FRET occurred when the complex between (CUG)12 RNA and MBNL1 formed, since the fluorophores are near each other. On the other hand, FRET does not occur in the presence of small-molecule inhibitors since the (CUG)12–MBNL1 interaction is disrupted. The TR-FRET screens identified inhibitors of the (CUG)12 RNA-MBNL1 protein complex exhibiting IC50 values at low micromolar level. One of the inhibitors improved DM1-associated pre-mRNA splicing defects in cell and mouse models of DM1 [Citation118].
FID is an assay that utilizes a fluorescently labeled indicator, which can be displaced upon ligand binding to the target molecule. FID has been used in primary HTS campaigns by many groups [Citation119]. The Hargrove group recently showed FID to be useful in identifying inhibitors of enterovirus 71 (EV71) viral translation and replication [Citation6]. In their study, the authors used a fluorescently labeled, highly basic peptide fragment as an indicator that can be displaced upon binding of small molecules to the EV71 stem loop II (SLII) IRES structure. The strongest binder to EV71 SLII, DMA-135, induces a conformational change in the RNA that stabilizes a ternary structure with the AU- rich element RNA-binding protein, thus inhibiting viral translation.
3.2. Affinity-based screening
A direct method for detecting RNA–small molecule complexes is electrospray ionization mass spectrometry (ESI-MS) [Citation120]. In an ESI-MS assay, the shift in the molecular weight of the unbound and unlabeled target upon the formation of the target–ligand complex is measured. ESI-MS was applied by Seth et al. [Citation97] to search for small molecules that bind to the IRES IIa subdomain of the hepatitis C virus (HCV). The authors identified benzimidazole inhibitors, which reduce viral RNA levels in the HCV replicon at low micromolar concentrations.
In indirect affinity-selection mass spectrometry (AS-MS) methods ()), small molecules that bind to the target are separated from the target prior to identification. One of the emerging indirect AS-MS assays is the automated ligand identification system (ALIS), which allows for the HTS of small molecules that bind to an unlabeled target. Originally used against protein targets, ALIS was adapted by Merck for use in the screening, ranking, and characterization of RNA binders [Citation21,Citation98,Citation121,Citation122]. In an ALIS assay, the target is first incubated with small-molecule compound mixtures. Then, the target–small-molecule complexes are purified by size exclusion chromatography, and the complexes are dissociated using reverse-phase chromatography. Finally, the bound small molecules are detected by MS. In seminal work by Rizvi et al. [Citation21], structurally diverse small molecules that bind to the FMN riboswitch were identified using the ALIS approach, and the binders induced a change in the riboswitch conformation different from that induced by the cognate FMN ligand. Very recently, Aguilar et al. [Citation98] applied ALIS to the study of the non-coding Xist RNA. They identified the drug-like compound, X1, with an indazole–benzimidazole scaffold to have submicromolar affinity to Xist RNA. By binding to Xist, X1 displaces interacting proteins, suppresses histone H3K27 trimethylation, and blocks initiation of X-chromosome inactivation.
3.3. Small molecule microarray (SMM)
SMM ()) has proven to be a robust and scalable method for discovering RNA-binding small molecules. In this method, a collection of small molecules is printed onto a glass surface using a robotic microarrayer [Citation123]. Then, the microarray is incubated with a fluorescently labeled target biomolecule and washed to remove unbound target. Next, the microarrays are imaged using a fluorescence scanner, and the fluorescence intensities of each spot on the microarray are quantified. Last, the spots with increased fluorescence upon incubation, representing interactions between the target and the small molecules, are revealed by statistical analysis.
The Schneekloth group used SMM as a platform for identifying RNA binders. In one example, they pooled a library of 20,000 drug-like compounds and incubated the library with Cy5-labeled HIV-1 TAR RNA (target) and a Cy5-labeled precursor of miR-21 hairpin (competitor) [Citation99]. After analysis of the microarray data, they identified a compound that selectively binds to TAR and does not resemble previously known TAR-binding small molecules. Further evaluation demonstrated that one of the analogues shows specificity for only the TAR region, lacks toxicity, and has good antiviral activity.
An ingenious variation on the standard SMM is the two-dimensional combinatorial screen (2DCS) developed by the Disney lab [Citation124]. 2DCS is a library-versus-library approach, wherein a small-molecule library conjugated onto an agarose-coated microarray is hybridized with a γ-32P-labeled RNA library, along with competitor oligonucleotides. 2DCS has enabled the assessment of millions of RNA–small-molecule interactions and the identification of novel RNA binders. The data generated by 2DCS has been incorporated into Inforna [Citation125,Citation126], which was used to identify binders of precursor microRNA-96. The binders inhibit the biogenesis of the microRNA, upregulate FOXO1, and induce apoptosis in cancer cells [Citation125].
3.4. Fragment-based screening
Fragment-based screening is gaining attention in recent years since it shows promise in developing RNA binders with drug-like properties [Citation127]. Briefly, fragment-based screening employs biophysical techniques to identify low molecular weight compounds. The fragments would have weak but specific interactions, but they may be optimized using a variety of strategies to obtain more potent and selective compounds.
Fragment-based screening was implemented to identify novel ligands for the E. coli thiamine pyrophosphate (TPP) thiM riboswitch [Citation128,Citation129], which is a regulatory element in the non-coding region of mRNA that controls gene expression by sensing TPP. A library of 1300 fragments was screened against the thiM riboswitch using a combination of equilibrium dialysis, NMR spectroscopy, and isothermal titration calorimetry. Structurally diverse fragments with equilibrium dissociation constant (Kd) values ranging from 22 to 670 µM were identified. The hit fragments were validated using X-ray crystallography, small-angle X-ray scattering, and SHAPE, which showed that the fragments compete with TPP but rearranges the riboswitch into a structure different from that of the cognate complex [Citation130].
Garavís et al. [Citation103] used 19F-NMR-based fragment screening ()) to identify small molecules that bind to telomeric repeat-containing RNAs (TERRAs), which fold into G-quadruplexes. TERRA G-quadruplexes act as scaffolds for the formation of telomeric heterochromatin in cancer cells and are thus ideal therapeutic targets [Citation131]. The screen identified 20 hit fragments from a 355-member fluorinated fragment library. Examination of the interactions of several of the hits suggested that the fragments favor the parallel propeller-like conformation of telomeric sequences.
3.5. DNA-encoded library (DEL) screening
DEL screening offers several advantages including the ability to screen extensive libraries, low cost, simplicity, and speed [Citation132]. A DEL is a collection of small-molecule compounds in which each compound is bound to a segment of synthetic DNA ()) [Citation133]. Each DNA segment serves as a barcode that contains information about the structure of the small molecule attached to it. DEL screening is an affinity-based process, which involves (1) incubation of the DEL compounds with the target, (2) removal of the weak or nonspecific binders, (3) sequencing of the DNA codes of the isolated binders, and (4) data analysis of the resulting DNA sequences to reveal the identities of the binders.
The Disney group recently reported the use of a strategy that integrates DEL with DCS to probe binding between RNAs and small molecules [Citation104]. They screened a 73,728-member DEL against 4,096 RNAs, thereby evaluating a total of ~300 million interactions. Using fluorescence-activated cell sorting, they identified both the DEL compounds that specifically bind to the RNAs and the selective RNAs. One of the notable findings of the study is a small molecule that binds with nanomolar affinity for primary microRNA-27a, inhibits miRNA biogenesis, and rescues a migratory phenotype in triple-negative breast cancer cells.
The biggest issue with DEL screening against RNA targets is that it could result in false positives due to the nonspecific binding of the DNA tags to the RNAs. In a recent report, HitGen circumvented this issue by utilizing RNA patches, competitive elution, and an algorithm to differentiate DNA–RNA binding from RNA–small-molecule binding [Citation105]. Using these strategies, they identified two compounds, from a 46.3 billion-member DEL, with low nanomolar affinity to FMN riboswitch and that compete with the cognate FMN ligand.
3.6. Virtual screening
Virtual screening can significantly reduce costs and expand the chemical space accessible, as well as remove inactive compounds prior to experimental testing. One of the virtual screening approaches involves the docking of virtual libraries to the 3D structure of the target, followed by evaluation of the binding energies using a scoring function ()). There are a few programs that were developed specifically for docking small molecules to RNA, such as MORDOR [Citation134] and rDock (formerly, RiboDock) [Citation135]. In addition, there are programs, originally designed for protein targets, that were adapted or reparameterized for small-molecule docking to RNA, including Dock6 [Citation136], ICM [Citation137], and AutoDock [Citation138].
Daldrop et al. [Citation107] used the program DOCK 3.5.54 to identify novel binders to the guanine riboswitch (GRA). First, they docked 23 compounds (test set)–8 known ligands and 15 decoys–to the crystal structure of the GRA carrying a C74U mutation [Citation139]. This resulted in a clear separation of the ligands and decoys. Then, they assembled a larger database containing 2,592 compounds plus the 23 compounds in the test set, docked the database to GRA, and ordered the compounds by score. A near perfect discrimination between known ligands and database compounds/decoys was obtained. Encouraged by these results, they performed more studies on the top scoring hits and found four new ligands of GRA with micromolar level binding affinities, two of which have scaffolds not resembling those of previously known ligands.
The Al-Hashimi group considered the large conformational changes in RNA targets by docking small molecules on RNA dynamic ensembles [Citation108]. The group first carried out 80 ns molecular dynamics (MD) simulations on NMR structures of HIV-1 TAR RNA to select 20 conformers that would make up the dynamic ensemble. Then, they subjected the TAR ensemble to virtual screening against ~51,000 small molecules. Fluorescence-based assays were performed using the top 57 hits, resulting in the identification of six compounds that bind to TAR that were not previously known to bind to TAR. One compound, netilmicin, exhibits exceptional selectivity for TAR and inhibits interaction with Tat and HIV replication.
3.7. Phenotypic screening
Using a mouse model for spinal muscular atrophy (SMA), PTC Therapeutics and Hoffmann-LaRoche performed phenotypic screening ()) to identify splicing modifiers of survival of motor neuron 2 (SMN2) with high selectivity and oral bioavailability [Citation113]. They employed a cell-based system in which the luciferase reporter protein was only expressed when exon 7 of the SMN2 minigene was included after splicing. Later studies revealed that the small molecules function by binding to the SMN2 pre-mRNA and the U1 snRNP [Citation140]. The SMN2 splicing modifiers were optimized, resulting in the discovery of the now FDA-approved risdiplam [Citation114].
Around the same time the PTC-Roche SMN2 splicing modifiers were identified, a team at Novartis also performed phenotypic screening to identify small molecules with the ability to reduce exon 7 exclusion [Citation115]. An NSC34 motor neuron cell line expressing an SMN2 minigene reporter was employed to screen their compound library (~1,400,000 compounds). Two active compounds – branaplam (originally NVS-SM1) and NVS-SM2 – were found to have good efficacy, bioavailability, and distribution to the brain. Branaplam is currently in a Phase 1/2 clinical trial in infants with SMA (NCT02268552) [Citation141]. Interestingly, branaplam also promotes the inclusion of a pseudoexon in huntingtin (HTT) transcripts, thereby downregulating HTT mRNAs and protein [Citation142].
A third example of the successful application of phenotypic screening in identifying RNA binders was reported by researchers at Merck, who screened a chemical library of ~57,000 compounds for antibacterial activity against an E. coli in the presence of riboflavin [Citation20]. They found that ribocil – a small molecule that does not resemble riboflavin – completely suppresses E. coli bioactivity. Later, they identified the target of ribocil to be the FMN riboswitch by selecting E. coli ribocil-resistant mutants and mapping ribocil-resistant mutations to their target using whole-genome sequencing. Further evaluation revealed that ribocil is a racemic mixture of isomers and that one of its enantiomers, ribocil-B, predominantly inhibits riboflavin biosynthesis and bacterial growth.
4. Biophysical techniques to confirm in vitro binding of the hit compounds
After screening for compounds that potentially bind the RNA targets, the next critical steps are: (1) to confirm that such compounds bind to the target, (2) assess the functional effects of the compounds, and (3) establish target engagement. In this section, several of the biophysical techniques used for confirming the binding of small molecules to an RNA target () will be described. To select the most appropriate technique, one should review the range, strengths, and limitations of each biophysical technique. Ideally, one should perform multiple biophysical assays in parallel or in a funnel format from the highest to the lowest throughput. Furthermore, appropriate experimental design and conditions, use of suitable controls, skill in the application of the technique, and correct and adequate data analysis are crucial for ensuring accurate and repeatable results. Given the multitude of biophysical techniques, this review will only cover several of the most powerful techniques for probing RNA–small-molecule interactions.
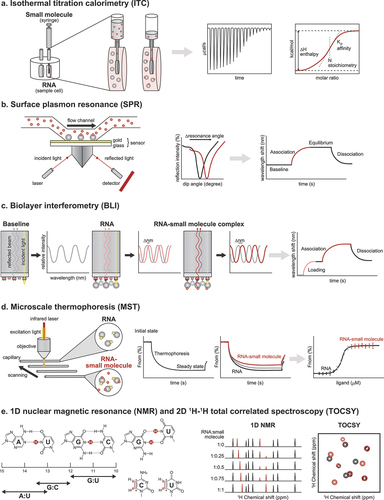
Figure 4. Biophysical techniques for confirming and validating hit compounds. (a) In an ITC experiment, the reference and sample cells should contain the same buffer because mismatched buffers lead to large background heats during the titration. Each peak in the raw data counts for one injection of the ligand solution, and the respective enthalpy is measured. The data is analyzed by fitting a sigmoidal curve to the released or absorbed enthalpy per mol plotted against the molar ratio. The binding affinity and stoichiometry can be determined from the fitted curve. (b) For SPR measurements, it is generally recommended to use low surface densities of the immobilized RNA and high ligand flow rate for accurate determination of kinetic constants. SPR causes a dip in the intensity of the reflected light at the sensor surface. A shift in the curve represents binding of the ligand to the RNA. An SPR sensogram can be divided into the association, steady state or equilibrium, and dissociation phases. (c) Since BLI experiments are conducted in uncovered plates and using small volumes, the entire experiment should be conducted swiftly (< 4 hours) to limit excessive evaporation, which can impact experimental results. In BLI, changes in the number of molecules bound to the biosensor tip shift the interference pattern that can be monitored in real-time. Association and dissociation are measured by dipping the sensor in solutions with or without the ligand, respectively. (d) Prior to MST assays, different assay conditions should be tested because the type of buffer, pH, and ionic strength can influence the thermophoretic movement of molecules and dramatically change the outcome. The RNA and ligand solutions are incubated and filled into a glass capillary. At a specific temperature, an infrared laser generates a temperature gradient, and RNA-small molecule complexes move slower in that gradient than unbound RNA. A binding curve can be generated from the difference between the fluorescence signals of the bound and unbound states, from which a binding constant can be derived. (e) The H1 and H3 protons of guanosines and uridines, respectively, as well as the H5–H6 crosspeaks of pyrimidines, can be perturbed after small molecule binding. By obtaining the 1H and 2D 1H −1H NMR spectra of the RNA target prior to and after the addition of small molecules, one can monitor these perturbations and determine if the small molecule binds to the RNA and which RNA residues bind the small molecule.
4.1. Isothermal titration calorimetry (ITC)
The gold standard for obtaining binding constants and thermodynamic parameters for target–ligand interactions is ITC [Citation143]. During an ITC experiment, a ligand (small molecule) solution is typically injected into a sample cell containing the label-free target biomolecule (RNA) ()). A sensitive calorimeter measures the heat released or absorbed during titration until the binding reaction has reached equilibrium. The measured heat values are then used to obtain the binding affinity (KD), stoichiometry (N), and change in enthalpy (∆H), from which the changes in free energy (∆G) and entropy (∆S) are calculated. Obtaining these thermodynamic parameters for binding of the hit compounds to the target RNA can help in compound classification and prioritization at a later stage. Although a trusted technique, ITC requires large amounts of sample and high solubility of the small-molecule compound.
4.2. Surface plasmon resonance (SPR)
Characterizing the kinetics of binding can lead to an understanding of the efficacy and safety of the small molecule and therefore enhance the success of the drug discovery program. SPR is the most extensively used technique for measuring the kinetics of small-molecule binding to an RNA [Citation144]. In an SPR experiment using a microfluidic system, the target biomolecule (RNA) is immobilized onto a sensor chip, and a pump is used to flow the liquid containing the ligand (small molecule) over the sensor chip ()). An optical system measures the changes in refractive index at the interface of the liquid and the sensor, which are plotted over time in the form of a sensogram. From the SPR experiment, one can obtain the kinetic parameters, such as the association and dissociation rate constants (kon and koff, respectively) and KD. SPR is highly sensitive, relatively high throughput, and has a small sample requirement. However, the target immobilized onto the sensor must be stable over time, and a high level of expertise and experience are necessary for carrying out a successful SPR experiment.
4.3. Biolayer interferometry (BLI)
Another optical-based technique, BLI, can be used to measure RNA–small-molecule interaction [Citation96,Citation145,Citation146]. BLI analyzes the interference patterns of white light reflected from the surface of a glass fiber biosensor ()). The biotinylated target (RNA) is first immobilized onto streptavidin biosensor tips, then the tips are dipped into a solution containing the ligand (small molecule). The interaction of the ligand with the target can be monitored by recording the changes in light interference, which directly correlate with changes in the thickness of the biolayer resulting from the association of the ligand. Like SPR, one can obtain the kon, koff, and KD values from a BLI experiment. Although SPR is widely used in the pharmaceutical industry, BLI is gaining popularity because it is cheaper and easier to use, enables significantly higher throughput, does not rely on fluidics, and is less technically demanding compared to SPR.
4.4. Microscale thermophoresis (MST)
One of the emerging biophysical techniques that is being applied for probing RNA–small-molecule interactions is MST [Citation147]. In an MST experiment, a capillary containing a fluorescently labeled molecule (the target RNA) and a nonfluorescent ligand (small molecule) is gently heated with an infrared laser to generate a temperature gradient, along which the molecule migrates in a phenomenon known as thermophoresis ()). The fluorescent molecule is mixed with different concentrations of the nonfluorescent ligand, and the effect of different concentrations of the ligand on the migration of the molecule is quantified to measure binding. While MST is versatile in its applications and does not require immobilization of the sample, preparing the samples may be time consuming and labeling may alter the binding properties of the small molecule. Moreover, MST does not provide the kinetic constants like SPR and BLI do and has a lower sensitivity than ITC.
4.5. Nuclear magnetic resonance (NMR)
RNA-observed NMR techniques provide a wealth of information regarding how a ligand interacts with its target RNA, and such information is extremely helpful for understanding the mechanism of recognition and rational design. The most straightforward method is chemical shift mapping with 1H NMR spectroscopy using the imino proton region, in which the perturbations in the solvent exchangeable H1 of guanosines and H3 of uridines can be monitored upon gradual addition of the ligand ()) [Citation148]. The H1 and H3 imino protons serve as hydrogen donors in base pairing interactions and are, thus, perturbed when a small molecule interacts with the RNA. Because of the distinct chemical shift ranges and minimal spectroscopic overlap, perturbations in these imino protons can be monitored in a simple 1D manner. One disadvantage of imino 1H NMR spectroscopy is that, because the unstructured imino protons cannot be observed, it cannot detect perturbations when they occur at the loops and bulges on the RNA. To monitor more changes in the RNA brought about by small-molecule binding, it is advisable to pair imino-based NMR characterizations with 2D 1H-1H total correlated spectroscopy (TOCSY, otherwise known as Homonuclear Hartmann-Hahn or HOHAHA). This method measures the H5–H6 cross-peak of pyrimidines ()), and since the H5 and H6 protons are non-solvent exchangeable and carbon-bound, TOCSY can robustly monitor perturbations even those of pyrimidines in unstructured loops and bulges.
5. Assessing the functional effects and target engagement of RNA-binding small molecules
As mentioned above, aside from determining the binding of small molecules to the RNA target with in vitro experiments, it is necessary to assess the functional and biochemical effects of the small molecules in a physiological context using cell-based assays. These assays can provide information about the efficacy, mechanism of action, cytotoxicity, cell permeability, stability, and off-target effects of the RNA binders. As is the case for protein-targeted small-molecule drug discovery, one must carefully select from a broad range of cell-based assay types and cell sources to simulate a physiological or disease-specific environment.
In addition, it is critical to assess whether the functional effect of the small molecule is a consequence of direct engagement of the RNA target. The techniques that have been used to assess engagement of small molecules to RNA targets are summarized in . It can be expected that drug regulatory authorities will expect a thorough investigation on the potential off-targets – both RNA and protein – of the small molecules especially since there are fewer examples of RNA-targeted small-molecule drugs.
Table 3. Techniques to assess engagement of small molecules to RNA targets.
6. Techniques for determining 3D structure
Although not a strict requirement in drug discovery, the 3D structure of the target–small-molecule complex helps us better understand how a small molecule binds to its target and gives us clues on how to optimize the small molecule for increased potency and selectivity. The three techniques for determining the 3D structures of RNA–small-molecule complexes are X-ray crystallography, NMR, and cryo-electron microscopy (cryo-EM) ().
Figure 5. Comparison of the techniques for the determination of the 3D structures of RNA–small-molecule complexes. (a) X-ray crystallography can provide atomic resolution structures for RNAs of any size but requires a large amount of sample that must be crystallized. The crystal structure of the FMN riboswitch in complex with the compound, BRX1151 (red), is shown [Citation22]. (b) Unlike X-ray crystallography, solution-state NMR structures provide information about the dynamics of the RNA target, but it also requires large amounts of sample and is not suited for solving the 3D structures of large RNAs. The solution structure of the UGGAA-UGGAA RNA pentad in complex with the naphthyridine carbamate dimer (NCD, red) is shown [Citation8]. (c) In recent years, the resolutions of cryo-EM structures of RNAs are improving, but it is still only suitable for large RNAs or RNA–protein complexes. Cryo-EM requires only a small amount of sample, and the obtained structures are closer to the RNA’s native state than the X-ray structures. The cryo-EM structure of the SAM IV riboswitch–SAM (red) complex is shown [Citation161].
![Figure 5. Comparison of the techniques for the determination of the 3D structures of RNA–small-molecule complexes. (a) X-ray crystallography can provide atomic resolution structures for RNAs of any size but requires a large amount of sample that must be crystallized. The crystal structure of the FMN riboswitch in complex with the compound, BRX1151 (red), is shown [Citation22]. (b) Unlike X-ray crystallography, solution-state NMR structures provide information about the dynamics of the RNA target, but it also requires large amounts of sample and is not suited for solving the 3D structures of large RNAs. The solution structure of the UGGAA-UGGAA RNA pentad in complex with the naphthyridine carbamate dimer (NCD, red) is shown [Citation8]. (c) In recent years, the resolutions of cryo-EM structures of RNAs are improving, but it is still only suitable for large RNAs or RNA–protein complexes. Cryo-EM requires only a small amount of sample, and the obtained structures are closer to the RNA’s native state than the X-ray structures. The cryo-EM structure of the SAM IV riboswitch–SAM (red) complex is shown [Citation161].](/cms/asset/3ca361a9-1942-4ce0-aec6-672e471712e3/iedc_a_2134852_f0005_oc.jpg)
6.1. X-ray crystallography
The first step in X-ray crystallography is obtaining single crystals of sufficient size and quality. After obtaining the crystal(s), a diffraction experiment is performed, where the crystal is illuminated with X-ray beams, producing a diffraction pattern. The crystal is gradually rotated to collect a set of diffraction patterns, which are converted into a 3D model that shows the electron density within the crystal. Based on the electron density map, the atomic model of the RNA–small-molecule complex is built and subsequently refined using computational tools for structure determination.
Over the past few decades, various crystallization strategies – the use of protein chaperones [Citation162], tetraloops [Citation163], kissing loops [Citation164], the tRNA scaffold [Citation165], and model RNAs [Citation166] – have improved the likelihood of obtaining crystals for RNA and its complexes. Moreover, crystallization robots have significantly reduced the amount of sample required for crystallization experiments and the experimental burden. The introduction of covalently or non-covalently bound heavy- or anomalously scattering atoms [Citation167–169] also led to successful crystallographic phase determination of RNAs. In addition, microfocus beamlines, detector technologies, and X-ray-free electron lasers enabled structure determination from substantially smaller crystals. Finally, the availability of highly automated and comprehensive computational systems, such as the Phenix software suite [Citation170], that can arrive at an initial model of a structure with minimal human intervention, has largely reduced the time consumed for structure determination.
X-ray crystallography can be used for RNAs of any size and provides atomic resolution images of the RNA–small-molecule structures ()). Although a powerful tool that delivers highly reliable structural information, X-ray crystallography requires that a sample be crystallizable, that the compound be soluble, and that a substantial percentage of binding sites be occupied by the ligand. In addition, an X-ray crystal structure represents only one (or at most, two) static form of the RNA–small-molecule complex.
6.2. Solution-state NMR
An NMR experiment is often performed on an RNA–small-molecule complex dissolved in sodium phosphate buffer with salt, usually sodium chloride. The water suppression technique allows measurement with a buffer containing 5–10% D2O without having to replace the buffer with 100% D2O. Prior to NMR experiments, it is advisable to test different annealing and salt conditions to determine the proper folding conditions for the RNA target. In some cases, the use of 13C/15N labeled RNA in multinuclear experiments can provide more chemical shift information and reduce signal overlap.
Typically, the 2D-NOESY spectra, focusing on the imino proton region, are first measured and the signals are assigned to determine the secondary structure [Citation171]. Then, NMR measurements (2D-NOESY or 2D 1H-1H TOCSY) for the non-exchangeable protons are performed, and the NOE cross-peaks between purine H2 (only adenine), H8, or pyrimidine H6 and pyrimidine H5 or ribose H1’ are assigned. Following signal assignment, structural information (proton–proton distance, dihedral angle, and base pairing) is derived, and a restraint file is created for structure calculations. Currently, most calculations are performed using the simulated annealing protocol, and the calculations are performed until the restraints are satisfied and the structure converges. Iterative rounds of signal assignment and structure calculations are made until an acceptable ensemble of structures is obtained.
RNA synthesis techniques have allowed the preparation of samples with different isotopic labels, which have proven to be useful in structure determination and dynamic studies [Citation172]. Furthermore, vast improvement in electronics and cryogenic probes increased the sensitivity of NMR measurements by tenfold over the past decade. Recently, Novakovic et al. proposed an approach for collecting NMR spectra for RNAs by injecting hyperpolarized water and reported a 300-fold enhancement of imino resonances [Citation173]. Other notable advances include computational approaches that improve the ability to probe the dynamic RNA ensembles, such as those for predicting chemical shifts from structure and generating conformational libraries from sequence [Citation174].
The distinguishing feature of solution-state NMR is that, as its name clearly implies, the 3D structure of RNA–small-molecule complex is measured in solution ()). Thus, NMR’s biggest advantage over X-ray crystallography is that it provides information about the dynamics of small-molecule binding. However, the NMR spectra of large RNAs may be very complicated and impossible to interpret, thereby limiting the application of NMR to smaller RNAs.
6.3. Cryo-EM
The continuing advances in cryo-EM offer unprecedented opportunities to visualize large RNA–small-molecule complexes. In a typical cryo-EM experiment, the samples are applied to a grid mesh and plunge frozen in liquid ethane in a process called vitrification. During this process, the sample solution is kept in an amorphous, noncrystalline state. The frozen grids are then screened for appropriate ice thickness, sample density, and particle orientation. Next, a series of two-dimensional images (micrographs) of the sample are taken, and computational tools are used to extract and average representative views of the molecules from the micrographs. In the end, atomic models are built into the EM maps and refined to generate the macromolecular structure.
Compared to X-ray crystallography and NMR, cryo-EM requires only a small amount of sample and can tolerate lower degrees of purity ()). Moreover, since there is no need to crystallize the sample, cryo-EM is more straightforward and provides structures that are closer to its native state than X-ray crystallography. However, cryo-EM structures of RNAs are rare, and the rare structures are only at a moderate resolution (3.0–10 Å). This is due to the intrinsic heterogeneity and small size of RNAs and the lack of effective approaches to obtain properly folded, stable RNAs. Efforts to overcome these challenges are underway and include: (1) strategies to create larger and more stable RNA nanoarchitectures [Citation175], (2) development of the Ribosolve workflow [Citation176] that integrates native gel analysis, mutate-and-map by next-generation sequencing, cryo-EM, and auto-DRRAFTER RNA modeling [Citation177], and (3) hybrid approaches combining RNA modeling, X-ray crystallography, and NMR.
7. Optimization of RNA-binding small molecules
Understanding the key interactions between an RNA target and a small molecule is highly beneficial for optimizing small molecules to improve their potency and selectivity. However, even when the 3D structure of an RNA–small-molecule complex is available, visual inspection is not enough to fully understand the molecular interactions. One of the promising methods for exploring molecular interactions is the fragment molecular orbital (FMO) method [Citation178], which provides a comprehensive list of the interactions and their strengths in terms of energy stabilization of the complex. FMO is much faster than traditional quantum mechanics (QM) methods because FMO calculations are done by partitioning a large system into fragments.
Recently, the Frank group applied the FMO method to probe the interactions between the FMN riboswitch and nine small molecules [Citation179]. Their analyses resulted in the identification of the hot-spot residues within the binding pocket on the FMN riboswitch, as well as in the confirmation of the dominance of electrostatic interaction in driving ligand binding. By examining the structure–energy relationships within the nine complexes, the group was also able to uncover an atypical T-shaped contact between a riboswitch residue and the ligands, which may further stabilize complex formation.
Even without comprehensive FMO analysis, one can still design potent and selective RNA-binding small molecules using the available 3D structure information, albeit with increased complexity. Vicens et al. [Citation22] used the structure-based drug design strategy to optimize roseoflavin mononucleotide, which binds in a nonspecific manner to the FMN riboswitch. In their study, they examined the 3D structures of complexes formed by FMN riboswitches to determine the key interactions that would need to be maintained or replaced in the analogues. Through careful dissection of the effects of the modifications on binding affinity and activity using a combination of chemical probing and transcription termination assays, they discovered 5FDQD, which binds to the FMN riboswitch and shows antibacterial activity.
8. Conclusion
Here, the different technologies for identifying therapeutically compelling RNA motifs, screening for small-molecule compounds that bind to them, confirming in vitro binding, assessing the bioactivity and establishing target engagement, determining the 3D structures of RNA–small-molecule complexes, and optimizing the hit compounds to identify lead molecules with increased potency and selectivity have been discussed. The RNA community has successfully been able to repurpose the experimental techniques and adapt the computational tools that were protein-centric for use in RNA targets, as well as create technologies tailored for RNA. As more advances in both experimental and computational technologies are made and leveraged to their fullest potential, we are likely to witness an emergence of more RNA-targeted small molecules drugs, particularly for diseases with unmet medical needs.
9. Expert opinion
Small molecules have been the preferred modality for drug development because of their good oral bioavailability, broad tissue distribution, lower risk of immunogenicity, and lower manufacturing cost. However, the pharmaceutical industry’s productivity has been rapidly declining due to the diminishing pool of disease-related proteins that are the conventional targets of small-molecule drug discovery. Thus, finding novel therapeutic targets is critical. Targeting RNAs with small molecules is one way to address this problem. There are already several encouraging evidence that RNA-targeted small-molecule drugs could be developed.
Nevertheless, there remain areas for further improvement. The first is in target identification. For example, the nearest neighbor parameters that are the core of RNA secondary structure prediction algorithms have changed only modestly since they were introduced by Xia et al. [Citation58] in 1998. More thermodynamic data – in a wide variety of RNA lengths, base pair arrangements, and physical conditions – plus a more rational way to consolidate the data are needed to achieve more accurate RNA secondary structure predictions [Citation180]. In addition, although some RNA secondary structure prediction software can predict non-Watson–Crick pairs and higher-order structures, such as pseudoknots and G-quadruplexes, none of them can predict all or with great accuracy. Also, while the chemical probing techniques have greatly improved since their infancy, it is still difficult to obtain accurate reactivity profiles. Therefore, secondary structure predictions should be coupled with rigorous experimental verification, much like what Vicens and Kieft proposed in their paper [Citation181]. Various groups, including ours, are developing technologies to overcome the above-mentioned limitations in target identification technologies. We can anticipate that RNA targets will be better characterized in the next 5 years.
A second area that requires improvement is the way we approach screening. With our predilection for drug-like small molecules, we tend to screen against RNA molecules with complex structures. There is a danger to this – we may preclude opportunities to discover drugs for diseases linked to RNAs with simpler structures. Repeat sequences that form stem loop structures are a good case in point. While we should continue our pursuit of drug-like modulators of RNA, we should in parallel venture outside the Lipinski space by screening with more diverse chemical libraries. Along with trying different chemical space, should resources allow, it would be beneficial to screen against a variety of RNA motifs using one chemical library, a strategy previously employed with 2DCS [Citation124] and DEL screening [Citation104]. This not only helps in weeding out promiscuous RNA binders but also in the comprehensive characterization of compound libraries.
The third area for improvement is in determining the 3D structures of RNA and RNA–small-molecule complexes. While many efforts have been made, there is still a dearth of RNA 3D structures; less than 1% of the total number of structures in the Protein Data Bank is for RNA. A new machine learning approach called Atomic Rotationally Equivariant Scorer (ARES) has been shown to effectively learn even from a small amount of structural data and outperform other 3D structure prediction software [Citation182]. This is an encouraging development, but ARES’ accuracy in predictions for RNAs is lower than that of AlphaFold for proteins. To improve its accuracy, we should obtain more 3D structure information using experimental methods to procure a larger data set on which to train ARES and other machine learning tools.
The fourth and last area that would benefit from more research is hit-to-lead optimization. A structure-based computer-assisted manner of optimizing compounds is ideal but has yet to be proven effective. Since even ‘structured RNAs’ populate an ensemble of conformations, it is possible that one compound may capture one conformation from the ensemble, and another compound (even an analogue) may capture another conformation. One way to address this situation is molecular dynamics (MD). Provided they are carefully applied, MD simulations on available 3D structures can be used to explore RNA dynamics and conformational changes on the submicrosecond time scale [Citation183]. Moreover, combining MD simulations with QM methods, such as the FMO method, would enable the evaluation of interaction energies between the RNA in its various forms and the hit compound, and that information is expected to guide drug design.
There remains an abundance of functional RNAs that are yet to be discovered. Comparative structure analyses have revealed conserved RNA structures that perform a certain function. Analyses of interactions with RNA-binding proteins hint at the functions of the previously uncharacterized RNA motifs they bind to. However, these analyses must be supported by experimental evidence. Moreover, the number of databases for RNA is continuing to grow, and this could cause confusion even to people in the RNA community. Thankfully, there are efforts being put forth to unify the data; RNAcentral is one of them [Citation184,Citation185]. Launched in 2014, RNAcentral is a comprehensive database containing information on non-coding RNAs spanning a broad range of functions and species. It is our hope that this database be expanded to also cover mRNA and pre-mRNA sequences that may have functions outside coding for proteins.
Article highlights
A variety of experimental and computational technologies have been developed for the discovery of RNA-targeted small molecules
Therapeutically valuable RNA motifs can be identified using in silico secondary structure prediction and chemical probing methods, but these methods still require further improvement for greater accuracy
The utility of target-based and phenotypic screening approaches in the discovery of RNA-targeted small molecules has been demonstrated against a wide variety of diseases
The binding of small-molecule compounds to the RNA target can be confirmed using in vitro biophysical techniques that are also commonly used for protein targets
After confirming binding, it is crucial to determine the functional and biochemical effects of the RNA-targeted small molecules, establish target engagement, and obtain three-dimensional structure information
This box summarizes key points contained in the article.
Declaration of interest
E Morishita is a senior investigator at Veritas In Silico. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgements
The author is grateful to Gota Kawai, Shingo Nakamura, Takashi Kamimura, Amiu Shino, and Maina Otsu for their helpful feedback and comments.
Additional information
Funding
References
- Pfeffer CG. The biotechnology sector: therapeutics. In: Burns LR, editor. The business of healthcare innovation. Cambridge (UK): Cambridge University Press; 2020. p. 89–302.
- Hewitt WM, Calabrese DR, Schneekloth JS. Evidence for ligandable sites in structured RNA throughout the Protein Data Bank. Bioorg Med Chem. 2019;27(11):2253–2260.
- Serganov A, Huang L, Patel DJ. Coenzyme recognition and gene regulation by a flavin mononucleotide riboswitch. Nature. 2009;458(7235):233–237.
- Stagno JR, Liu Y, Bhandari YR, et al. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature. 2017;541(7636):242–246.
- Michiels PJ, Versleijen AA, Verlaan PW, et al. Solution structure of the pseudoknot of SRV-1 RNA, involved in ribosomal frameshifting. J Mol Biol. 2001;310(5):1109–1123.
- Davila-Calderon J, Patwardhan NN, Chiu L-Y, et al. IRES-targeting small molecule inhibits enterovirus 71 replication via allosteric stabilization of a ternary complex. Nat Commun. 2020;11(1):4775.
- Faber C, Sticht H, Schweimer K, et al. Structural rearrangements of HIV-1 Tat- responsive RNA upon binding of neomycin B. J Biol Chem. 2000;275(27):20660–20666.
- Shibata T, Nagano K, Ueyama M, et al. Small molecule targeting r(UGGAA)n disrupts RNA foci and alleviates disease phenotype in Drosophila model. Nat Commun. 2021;12(1):236.
- Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009;10(1):168.
- Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. 2016;16(3):167–179.
- ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816.
- Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327(6121):389–394.
- Wong CH, Hendrix M, Priestley ES, et al. Specificity of aminoglycoside antibiotics for the A-site of the decoding region of ribosomal RNA. Chem Biol. 1998;5(7):397–406.
- Verhelst SH, Michiels PJ, vander Marel GA, et al. Surface plasmon resonance evaluation of various aminoglycoside-RNA hairpin interactions reveals low degree of selectivity. Chembiochem. 2004;5(7):937–979.
- Morgan BS, Forte JE, Culver RN, et al. Discovery of key physicochemical, structural, and spatial properties of RNA-targeted bioactive ligands. Angew Chem Int Ed Engl. 2017;56(43):13498–13502.
- Donlic A, Swanson EG, Chiu LY, et al. R-BIND 2.0: an updated database of bioactive RNA-targeting small molecules and associated RNA secondary structures. ACS Chem Biol. 2022;17(6):1556–1566.
- Warner KD, Hajdin CE, Weeks KM. Principles for targeting RNA with drug-like small molecules. Nat Rev Drug Discov. 2018;17(8):547–558.
- Juru U, Hargrove A, A E. Frameworks for targeting RNA with small molecules. J Biol Chem. 2020;295(1):296.
- Chavali SS, Bonn-Breach R, Wedekind JE. Face-time with TAR: portraits of an HIV-1 RNA with diverse modes of effector recognition relevant for drug discovery. J Biol Chem. 2019;294(24):9326–9341.
- Howe JA, Wang H, Fischmann TO, et al. Selective small-molecule inhibition of an RNA structural element. Nature. 2015;526(7575):672–677.
- Rizvi NF, Howe JA, Nahvi A, et al. Discovery of selective RNA-binding small molecules by affinity-selection mass spectrometry. ACS Chem Biol. 2018;13(3):820–831.
- Vicens Q, Mondragón E, Reyes FE, et al. Structure-activity relationship of flavin analogues that target the flavin mononucleotide riboswitch. ACS Chem Biol. 2018;13(10):2908–2919.
- Park SJ, Kim YG, Park HJ. Identification of RNA pseudoknot-binding ligand that inhibits the −1 ribosomal frameshifting of SARS-coronavirus by structure-based virtual screening. J Am Chem Soc. 2011;133(26):10094–10100.
- Davis DR, Seth PP. Therapeutic targeting of HCV internal ribosomal entry site RNA. Antivir Chem Chemother. 2011;21(3):117–128.
- Disney MD, Dwyer BG, Childs-Disney JL. Drugging the RNA World. Cold Spring Harb Perspect Biol. 2018;10(11):a034769.
- Verma AK, Khan E, Bhagwat SR, et al. Exploring the potential of small molecule-based therapeutic approaches for targeting trinucleotide repeat disorders. Mol Neurobiol. 2009;57:566–584.
- Hamada M, Kiryu H, Sato K, et al. Prediction of RNA secondary structure using generalized centroid estimators. Bioinformatics. 2009;25(4):465–473.
- Sato K, Hamada M, Asai K, et al. CENTROIDFOLD: a web server for RNA secondary structure prediction. Nucleic Acids Res. 2009;37(Web Server):W277–80.
- Hamada M, Sato K, Kiryu H, et al. Predictions of RNA secondary structure by combining homologous sequence information. Bioinformatics. 2009;25(12):i330–8.
- Zakov S, Goldberg Y, Elhadad M, et al. Rich parameterization improves RNA structure prediction. J Comput Biol. 2011;18(11):1525–1542.
- Do CB, Woods DA, Batzoglou S, et al. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics. 2006;15(14):e90–8.
- Bleckley S, Stone JW, Schroeder SJ. Crumple: a method for complete enumeration of all possible pseudoknot-free RNA secondary structures. PLoS One. 2012;7(12):e52414.
- Bindewald E, Kluth T, Shapiro BA. CyloFold: secondary structure prediction including pseudoknots. Nucleic Acids Res. 2010;38(Web Server):W368–72.
- Swenson MS, Anderson J, Ash A, et al. GTfold: enabling parallel RNA secondary structure prediction on multi-core desktops. BMC Res Notes. 2012;5(1):341.
- Sato K, Kato Y, Hamada M, et al. IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics. 2011;27(13):i85–93.
- Xayaphoummine A, Bucher T, Thalmann F, et al. Prediction and statistics of pseudoknots in RNA structures using exactly clustered stochastic simulations. Proc Natl Acad Sci. 2003;100(26):15310–15315.
- Xayaphoummine A, Bucher T, Isambert H. Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Res. 2005;33(suppl_2):W605–10.
- Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452(7183):51–55.
- Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9(1):133–148.
- Rivas E, Eddy SR. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J Mol Biol. 1999;285(5):2053–2068.
- Reeder J, Steffen P, Pknotsrg GR. pknotsRG: RNA pseudoknot folding including near-optimal structures and sliding windows. Nucleic Acids Res. 2007;35:W320–4.
- Gruber AR, Lorenz R, Bernhart SH, et al. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:W70–4. DOI:10.1093/nar/gkn188.
- Hofacker IL, Stadler PF. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics. 2006;22(10):1172–1176.
- Giegerich R, Voss B, Rehmsmeier M. Abstract shapes of RNA. Nucleic Acids Res. 2004;32(16):4843–4851.
- Voss B, Giegerich R, Rehmsmeier M. Complete probabilistic analysis of RNA shapes. BMC Biol. 2006;4(1):5.
- Mathews DH, Disney MD, Childs JL, et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci. 2004;101(19):7287–7292.
- Mathews DH. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA. 2004;10(8):1178–1190.
- Tsang HH, Wiese KC. SARNA-Predict: accuracy improvement of RNA secondary structure prediction using permutation-based simulated annealing. IEEE/ACM Trans Comput Biol Bioinform. 2010;7(4):727–740.
- Ding Y, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res. 2003;31(24):7280–7301.
- Schroeder SJ, Stone JW, Bleckley S, et al. Ensemble of secondary structures for encapsidated satellite tobacco mosaic virus RNA consistent with chemical probing and crystallography constraints. Biophys J. 2011;101(1):167–175.
- Singh J, Hanson J, Paliwal K, et al. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nat Commun. 2019;10(1):5407.
- Barsacchi M, Novoa EM, Kellis M, et al. SwiSpot: modeling riboswitches by spotting out switching sequences. Bioinformatics. 2016;32(21):3252–3259.
- Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol. 2008;453:3–31.
- Dawson W, Fujiwara K, Kawai G, et al. A method for finding optimal RNA secondary structures using a new entropy model (vsfold). Nucleosides Nucleotides Nucleic Acids. 2006;25(2):171–189.
- Dawson WK, Fujiwara K, Kawai G. Prediction of RNA pseudoknots using heuristic modelling with mapping and sequential folding. PLoS One. 2007;2:e905.
- Dawson W, Takai T, Ito N, et al. A new entropy model for RNA: part III. Is the folding free energy landscape of RNA funnel shaped? J Nucleic Acids Invest. 2014;5(1):2652.
- Mathews DH, Turner DH. Prediction of RNA secondary structure by free energy minimization. Curr Opin Struct Biol. 2006;16(3):270–278.
- Xia T, Santalucia J, Burkard ME, et al. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry. 1998;37(42):14719–14735.
- Turner DH, Mathews DH. NNDB: the nearest neighbor parameter database for predicting stability of nucleic acid secondary structure. Nucleic Acids Res. 2009;38(suppl_1):D280–2.
- Mathews DH, Sabina J, Zuker M, et al. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288(5):911–940.
- Woese CR, Pace NR. Probing RNA structure, function, and history by comparative analysis. In: Gesteland RF, Atkins JF, editors. RNA World. New York (NY): Cold Spring Harbor Press; 1993. p. 91–117.
- Gutell RR, Lee JC, Cannone JJ. The accuracy of ribosomal RNA comparative structure models. Curr Opin Struct Biol. 2002;12(3):301–310.
- Moazed D, Stern S, Noller HF. Rapid chemical probing of conformation in 16S ribosomal RNA and 30S ribosomal subunits using primer extension. J Mol Biol. 1986;187(3):399–416.
- Merino EJ, Wilkinson KA, Coughlan JL, et al. RNA structure analysis at single nucleotide resolution by selective 2’-hydroxyl acylation and primer extension (SHAPE). J Am Chem Soc. 2005;127(12):4223–4231.
- Mortimer SA, Weeks KM. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J Am Chem Soc. 2007;129(14):4144–4145.
- Jackson TJ, Spriggs RV, Burgoyne NJ, et al. Evaluating bias-reducing protocols for RNA sequencing library preparation. BMC Genomics. 2014;15(1):569.
- Fuchs RT, Sun Z, Zhuang F, et al. Bias in ligation-based small RNA sequencing library construction is determined by adaptor and RNA structure. PLoS One. 2015;10(5):e0126049.
- Zubradt M, Gupta P, Persad S, et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat Methods. 2017;14(1):75–82.
- Siegfried NA, Busan S, Rice GM, et al. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat Methods. 2014;11(9):959–965.
- Smola MJ, Rice GM, Busan S, et al. Selective 2’-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat Protoc. 2015;10:1643–1669.
- Busan S, Weeks KM. Accurate detection of chemical modifications in RNA by mutational profiling (MaP) with ShapeMapper 2. RNA. 2017;24(2):143–148.
- Nakamura S, Jin L, Shino A, et al., inventors; Veritas In Silico Inc., assignee. Method for screening compound for controlling RNA function. WIPO (PCT) patent W0 2019/177103 A1.
- Andrews RJ, Roche J, Moss WN. ScanFold: an approach for genome-wide discovery of local RNA structural elements—applications to Zika virus and HIV. PeerJ. 2018;6:e6136.
- Davuluri RV, Suzuki Y, Sugano S, et al. CART classification of human 5‘ UTR sequences. Genome Res. 2000;10(11):1807–1816.
- Hentze MW, Caughman SW, Rouault TA, et al. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science. 1987;238(4833):1570–1573.
- Kumari S, Bugaut A, Huppert J, et al. An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nat Chem Biol. 2007;3(4):218–221.
- Cohen-Chalamish S, Hasson A, Weinberg D, et al. Dynamic refolding of IFN-gamma mRNA enables it to function as PKR activator and translation template. Nat Chem Biol. 2009;5(12):896–903.
- Mailliot J, Martin F. Viral internal ribosomal entry sites: four classes for one goal. Wiley Interdiscip Rev RNA. 2017;9(2). DOI:10.1002/wrna.1458
- Komar AA, Hatzoglou M. Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle. 2011;10(2):229–240.
- Estes PA, Cooke NE, Liebhaber SA. A native RNA secondary structure controls alternative splice-site selection and generates two human growth hormone isoforms. J Biol Chem. 1992;267(21):14902–14908.
- Varani L, Hasegawa M, Spillantini MG, et al. Structure of tau exon 10 splicing regulatory element RNA and destabilization by mutations of frontotemporal dementia and parkinsonism linked to chromosome 17. Proc Natl Acad Sci. 1999;96(14):8229–8234.
- Singh NN, Singh RN, Androphy EJ. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 2007;35(2):371–389.
- Brook JD, Mccurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell. 1992;68(4):799–808.
- Holden KL, Harris E. Enhancement of dengue virus translation: role of the 3’ untrans lated region and the terminal 3’ stem-loop domain. Virology. 2004;329(1):119–133.
- Jiang H, Mankodi A, Swanson MS, et al. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13(24):3079–3088.
- Yuan Y, Compton SA, Sobczak K, et al. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 2007;35(16):5474–5486.
- Gu W, Zhou T, Wilke CO, et al. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput Biol. 2010;6(2):e1000664.
- Joazeiro C. Mechanisms and functions of ribosome-associated protein quality control. Nat Rev Mol Cell Biol. 2019;20(6):368–383.
- Bell NM, L’Hernault A, Murat P, et al. Targeting RNA-protein interactions within the human immunodeficiency virus type 1 lifecycle. Biochemistry. 2013;52(51):9269–9274.
- Zhang Y, Zeng D, Cao J, et al. Interaction of quindoline derivative with telomeric repeat-containing RNA induces telomeric DNA-damage response in cancer cells through inhibition of telomeric repeat factor 2. Biochim Biophys Acta Gen Subj. 2017;1861(12):3246–3256.
- Fedorova O, GE J Jr, Adams RL, et al. Small molecules that target group II introns are potent antifungal agents. Nat Chem Biol. 2018;14(12):1073–1078.
- Simone R, Balendra R, Moens TG, et al. G-quadruplex-binding small molecules ameliorate C9orf72 FTD / ALS pathology in vitro and in vivo. EMBO Mol Med. 2018;10(1):22–31.
- Rzuczek SG, Southern MR, Disney MD. Studying a drug-like, RNA-focused small molecule library identifies compounds that inhibit RNA toxicity in myotonic dystrophy. ACS Chem Biol. 2015;10(12):2706–2715.
- Su Z, Zhang Y, Gendron TF, et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron. 83(5):1043–1050.
- Garcia-Lopez A, Tessaro F, Jonker HRA, et al. Targeting RNA structure in SMN2 reverses spinal muscular atrophy molecular phenotypes. Nat Commun. 2018;9(1):2032.
- Wang ZF, Ursu A, Childs-Disney JL, et al. The hairpin form of r(G4C2)exp in c9ALS/FTD is repeat-associated non-ATG translated and a target for bioactive small molecules. Cell Chem Biol. 2019;26(2):179–190.
- Seth PP, Miyaji A, Jefferson EA, et al. SAR by MS: discovery of a new class of RNA-binding small molecules for the hepatitis C virus: internal ribosome entry site IIA subdomain. J Med Chem. 2005;48(23):7099–7102.
- Aguilar R, Spencer KB, Kesner B, et al. Targeting Xist with compounds that disrupt RNA structure and X inactivation. Nature. 2022;604(7904):160–166.
- Sztuba-Solinska J, Shenoy SR, Gareiss P, et al. Identification of biologically active, HIV TAR RNA-binding small molecules using small molecule microarrays. J Am Chem Soc. 2014;136(23):8402–8410.
- Abulwerdi FA, Xu W, Ageeli AA, et al. Selective small-molecule targeting of a triple helix encoded by the long noncoding RNA, MALAT1. ACS Chem Biol. 2019;14(2):223–235.
- Childs-Disney JL, Tran T, Vummidi BR, et al. A massively parallel selection of small molecule-RNA motif binding partners informs design of an antiviral from sequence. Chem. 2018;4(10):2384–2404.
- Lee MK, Bottini A, Kim M, et al. A novel small-molecule binds to the influenza A virus RNA promoter and inhibits viral replication. Chem Commun (Camb). 2013;50(3):368–370.
- Garavís M, López-Méndez B, Somoza A, et al. Discovery of selective ligands for telomeric RNA G-quadruplexes (TERRA) through 19 F-NMR based fragment screening. ACS Chem Biol. 2014;18(7):1559–1566.
- Benhamou RI, Suresh BM, Tong Y, et al. DNA-encoded library versus RNA-encoded library selection enables design of an oncogenic noncoding RNA inhibitor. Proc Natl Acad Sci. 2022;119(6):e2114971119.
- Chen Q, Li Y, Lin C, et al. Expanding the DNA-encoded library toolbox: identifying small molecules targeting RNA. Nucleic Acids Res. 2022;50:gkac173.
- Lind KE, Du Z, Fujinaga K, et al. Structure-based computational database screening, in vitro assay, and NMR assessment of compounds that target TAR RNA. Chem Biol. 2002;9(2):185–193.
- Daldrop P, Reyes FE, Robinson DA, et al. Novel ligands for a purine riboswitch discovered by RNA-ligand docking. Chem Biol. 2011;18(3):324–335.
- Stelzer AC, Frank AT, Kratz JD, et al. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat Chem Biol. 2011;7(8):553–559.
- González-Bulnes L, Ibáñez I, Bedoya LM, et al. Structure-based design of an RNA-binding p -terphenylene scaffold that inhibits HIV-1 rev protein function. Angew Chem Int Ed Engl. 2013;52(50):13405–13409.
- Nguyen L, Luu LM, Peng S, et al. Rationally designed small molecules that target both the DNA and RNA causing myotonic dystrophy type 1. J Am Chem Soc. 2015;137(44):14180–14189.
- González ÀL, Konieczny P, Llamusi B, et al. In silico discovery of substituted pyrido[2,3-d]pyrimidines and pentamidine-like compounds with biological activity in myotonic dystrophy models. PLoS One. 2017;12(6):e0178931.
- Ren Y, Wang YF, Zhang J, et al. Targeted design and identification of AC1NOD4Q to block activity of HOTAIR by abrogating the scaffold interaction with EZH2. Clin Epigenetics. 2019;11(1):29.
- Naryshkin NA, Weetall M, Dakka A, et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345:688–693.
- Ratni H, Ebeling M, Baird J, et al., Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem. 2018;61(15): 6501–6517.
- Palacino J, Swalley SE, Song C, et al., SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat Chem Biol. 2015;11(7): 511–517.
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256.
- Chen CZ, Sobczak K, Hoskins J, et al. Two high-throughput screening assays for aberrant RNA-protein interactions in myotonic dystrophy type 1. Anal Bioanal Chem. 2012;402(5):1889–1898.
- Parkesh R, Childs-Disney JL, Nakamori M, et al. Design of a bioactive small molecule that targets the myotonic dystrophy type 1 RNA via an RNA motif-ligand database and chemical similarity searching. J Am Chem Soc. 2012;134:4731–4742.
- Wicks SL, Hargrove AE. Fluorescent indicator displacement assays to identify and characterize small molecule interactions with RNA. Methods. 2019;167:3–14.
- Dremann DN, Chow CS. The use of electrospray ionization mass spectrometry to monitor RNA-ligand interactions. Methods Enzymol. 2019;623:315–337.
- Rizvi NF, Nickbarg EB. RNA-ALIS: methodology for screening soluble RNAs as small molecule targets using ALIS affinity-selection mass spectrometry. Methods. 2019;167:28–38.
- Rizvi NF, Maria JP, Nahvi A, et al. Targeting RNA with small molecules: identification of selective, RNA-binding small molecules occupying drug-like chemical space. SLAS Discov. 2020;25(4):384–396.
- Connelly CM, Abulwerdi FA, Schneekloth JS. Discovery of RNA binding small molecules using small molecule microarrays. Methods Mol Biol. 2017;1518:157–175.
- Disney MD, Labuda LP, Paul DJ, et al., Two-dimensional combinatorial screening identifies specific aminoglycoside-RNA internal loop partners. J Am Chem Soc. 2008;130(33): 11185–11194.
- Velagapudi SP, Gallo SM, Disney MD. Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat Chem Biol. 2014;10(4):291–297.
- Disney MD, Winkelsas AM, Velagapudi SP, et al. Inforna 2.0: a platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem Biol. 2016;11(6):1720–1728.
- Lundquist KP, Panchal V, Gotfredsen CH, et al. Fragment-based drug discovery for RNA targets. Chem Med Chem. 2021;16(17):2588–2603.
- Chen L, Cressina E, Leeper FJ, et al. A fragment-based approach to identifying ligands for riboswitches. ACS Chem Biol. 2010;5:355–363.
- Cressina E, Chen L, Moulin M, et al. Identification of novel ligands for thiamine pyrophosphate (TPP) riboswitches. Biochem Soc Trans. 2011;39(2):652–657.
- Warner KD, Homan P, Weeks KM, et al. Validating fragment-based drug discovery for biological RNAs: lead fragments bind and remodel the TPP riboswitch specifically. Chem Biol. 2014;21(5):591–595.
- Bryan TM, Englezou A, Gupta J, et al. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995;14(17):4240–4248.
- Decurtins W, Wichert M, Franzini RM, et al. Automated screening for small organic ligands using DNA-encoded chemical libraries. Nat Protoc. 2016;11(4):764–780.
- Clark MA, Acharya RA, Arico-Muendel CC, et al. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat Chem Biol. 2009;5(9):647–654.
- Guilbert C, James TL. Docking to RNA via root-mean-square-deviation-driven energy minimization with flexible ligands and flexible targets. J Chem Inf Model. 2008;48(6):1257–1268.
- Ruiz-Carmona S, Alvarez-Garcia D, Foloppe N, et al. rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput Biol. 2014;10(4):e1003571.
- Lang PT, Brozell SR, Mukherjee S, et al. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA. 2009;15(6):1219–1230.
- Filikov AV, Mohan V, Vickers TA, et al. Identification of ligands for RNA targets via structure-based virtual screening: HIV-1 TAR. J Comput Aided Mol Des. 2000;14(6):593–610.
- Moitessier N, Westhof E, Hanessian S. Docking of aminoglycosides to hydrated and flexible RNA. J Med Chem. 2006;49(3):1023–1033.
- Gilbert SD, Reyes FE, Edwards AL, et al. Adaptive ligand binding by the purine riboswitch in the recognition of guanine and adenine analogs. Structure. 2009;17(6):857–868.
- Sivaramakrishnan M, Mccarthy KD, Campagne S, et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat Commun. 2017;8(1):1476.
- An open label study of LMI070 (branaplam) in type 1 spinal muscular atrophy [Internet]. Novartis Pharmaceuticals. clinicaltrials.gov. [ cited 2014 Oct 20]. Available from: https://clinicaltrials.gov/ct2/show/NCT02268552.
- Keller CG, Shin Y, Monteys AM. An orally available, brain penetrant, small molecule lowers huntingtin levels by enhancing pseudoexon inclusion. Nat Commun. 2022;13(1):1150.
- Kumar GS, Basu A. The use of calorimetry in the biophysical characterization of small molecule alkaloids binding to RNA structures. Biochim Biophys Acta. 2016;1860(5):930–944.
- Vo T, Paul A, Kumar A, et al. Biosensor-surface plasmon resonance: a strategy to help establish a new generation RNA-specific small molecules. Methods. 2019;167:15–27.
- Childs-Disney JL, Yildirim I, Park H, et al. Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity. ACS Chem Biol. 2014;9(2):538–550.
- Yang WY, He F, Strack RL, et al. Small molecule recognition and tools to study modulation of r(CGG) exp in Fragile X-associated tremor ataxia syndrome. ACS Chem Biol. 2016;11(9):2456–2465.
- Moon MH, Hilimire TA, Sanders AM, et al. Measuring RNA-ligand interactions with microscale thermophoresis. Biochemistry. 2018;57(31):4638–4643.
- Thompson RD, Baisden JT, Zhang Q. NMR characterization of RNA small molecule interactions. Methods. 2019;167:67–77.
- Guan L, Disney MD. Covalent small-molecule–RNA complex formation enables cellular profiling of small-molecule–RNA interactions. Angew Chem Int Ed Engl. 2013;52(38):10010–10013.
- Yang W-Y, Wilson HD, Velagapudi SP, et al. Inhibition of non-ATG translational events in cells via covalent small molecules targeting RNA. J Am Chem Soc. 2015;137(16):5336–5345.
- Velagapudi SP, Cameron MD, Haga CL, et al. Design of a small molecule against an oncogenic noncoding RNA. Proc Natl Acad Sci U S A. 2016;113:5898–5903.
- Rzuczek SG, Colgan LA, Nakai Y, et al. Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat Chem Biol. 2017;13(2):188–193.
- Costales MG, Haga CL, Velagapudi SP, et al. Small molecule inhibition of microRNA-210 reprograms an oncogenic hypoxic circuit. J Am Chem Soc. 2017;139(9):3446–3455.
- Velagapudi SP, Luo Y, Tran T, et al. Defining RNA-small molecule affinity landscapes enables design of a small molecule inhibitor of an oncogenic noncoding RNA. ACS Cent Sci. 2017;3(3):205–216.
- Costales MG, Hoch DG, Abegg D, et al. A designed small molecule inhibitor of a non-coding RNA sensitizes HER2 negative cancers to herceptin. J Am Chem Soc. 2019;141(7):2960–2974.
- Zhang P, Liu X, Abegg D, et al. Reprogramming of protein-targeted small-molecule medicines to RNA by ribonuclease recruitment. J Am Chem Soc. 2021;143(33):13044–13055.
- Tong Y, Gibaut QMR, Rouse W, et al. Transcriptome-wide mapping of small-molecule RNA-binding sites in cells informs an isoform-specific degrader of QSOX1 mRNA. J Am Chem Soc. 2022;144(26):11620–11625.
- Mukherjee H, Blain JC, Vandivier LE, et al. PEARL-seq: a photoaffinity platform for the analysis of small molecule-RNA interactions. ACS Chem Biol. 2020;15(9):2374–2381.
- Sexton AN, Vandivier LE, Petter JC, et al. Determination of RNA-ligand interactions with the photoaffinity platform PEARL-seq. Methods. 2022;205:83–88.
- Li Y, Disney MD. Precise small molecule degradation of a noncoding RNA identifies cellular binding sites and modulates an oncogenic phenotype. ACS Chem Biol. 2018;13(11):3065–3071.
- Zhang K, S L, Kappel K, et al. Cryo-EM structure of a 40 kDa SAM-IV riboswitch RNA at 3.7 Å resolution. Nat Commun. 2019;10(1):5511.
- Ferré-D’amaré AR. Use of the spliceosomal protein U1A to facilitate crystallization and structure determination of complex RNAs. Methods. 2010;52(2):159–167.
- Ferré-D’amaré AR, Zhou K, Doudna JA. A general module for RNA crystallization. J Mol Biol. 1998;279(3):621–631.
- Krummel DAP, Oubridge C, Leung AK, et al. Crystal structure of human spliceosomal U1 snRNP at 5.5 A resolution. Nature. 2009;458(7237):475–480.
- Lee E, Bujalowski PJ, Teramoto T. Structures of flavivirus RNA promoters suggest two binding modes with NS5 polymerase. Nat Commun. 2021;12(1):2530.
- Kondo J. Crystallographic studies of the ribosomal A-site molecular switches by using model RNA oligomers. Methods Mol Biol. 2016;1320:315–327.
- Correll CC, Freeborn B, Moore PB, et al. Use of chemically modified nucleotides to determine a 62-nucleotide RNA crystal structure: a survey of phosphorothioates, Br, Pt and Hg. J Biomol Struct Dyn. 1997;15(2):165–172.
- Baugh C, Grate D, Wilson C. 2.8 Å crystal structure of the malachite green aptamer. J Mol Biol. 2000;301(1):117–128.
- Lin L, Huang Z. Se-derivatized RNAs for X-ray crystallography. Methods Mol Biol. 2012;941:213–225.
- Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(2):213–221.
- Sakamoto T, Otsu M, Kawai G. NMR studies on RNA: The Nuclear Magnetic Resonance Society of Japan. Singapore: Springer; 2018. Experimental Approaches of NMR Spectroscopy. p. 439–459.
- Olenginski LT, Taiwo KM, Leblanc RM, et al. Isotope-labeled RNA building blocks for NMR structure and dynamics studies. Molecules. 2021;26(18):5581.
- Novakovic M, Olsen GL, Pintér G, et al. A 300-fold enhancement of imino nucleic acid resonances by hyperpolarized water provides a new window for probing RNA refolding by 1D and 2D NMR. Proc Natl Acad Sci. 2020;117(5):2449–2455.
- Liu B, Shi H, Al-Hashimi HM. Developments in solution-state NMR yield broader and deeper views of the dynamic ensembles of nucleic acids. Curr Opin Struct Biol. 2021;70:16–25.
- Liu D, Thélot FA, Piccirilli JA, et al. Sub-3-Å cryo-EM structure of RNA enabled by engineered homomeric self-assembly. Nat Methods. 2022;19(5):576–585.
- Kappel K, Zhang K, Su Z, et al. Accelerated cryo-EM-guided determination of three-dimensional RNA-only structures. Nat Methods. 2020;17(7):699–707.
- Kappel K, Liu S, Larsen KP, et al. De novo computational RNA modeling into cryo-EM maps of large ribonucleoprotein complexes. Nat Methods. 2018;15(11):947–954.
- Fukuzawa K, Tanaka S. Fragment molecular orbital calculations for biomolecules. Curr Opin Struct Biol. 2022;72:127–134.
- Deb I, Wong H, Tacubao C, et al. Quantum mechanics helps uncover atypical recognition features in the flavin mononucleotide riboswitch. J Phys Chem B. 2021;125(30):8342–8350.
- Dawson WK, Shino A, Kawai G, et al. Developing an updated strategy for estimating the free-energy parameters in RNA duplexes. Int J Mol Sci. 2021;22(18):9708.
- Vicens Q, Kieft JS. Thoughts on how to think (and talk) about RNA structure. Proc Natl Acad Sci. 2022;119(17):e2112677119.
- Townshend RJL, Eismann S, Watkins AM, et al. Geometric deep learning of RNA structure. Science. 2021;373(6558):1047–1051.
- Šponer J, Bussi G, Krepl M, et al. RNA structural dynamics as captured by molecular simulations: a comprehensive overview. Chem Rev. 2018;118(8):4177–4338.
- The RNAcentral Consortium, RNAcentral: a hub of information for non-coding RNA sequences. Nucleic Acids Res. 2019;47(D1):D221–9.
- Consortium R, Petrov AI, Ribas CE. RNAcentral 2021: secondary structure integration, improved sequence search and new member databases. Nucleic Acids Res. 2021;49(D1):D212–20.