
ABSTRACT
Started as an academic curiosity more than two decades ago, the idea that ion channels can regulate cellular processes in ways that do not depend on their conducting properties (non-ionic functions) gained traction and is now a flourishing area of research. Channels can regulate physiological processes including actin cytoskeletal remodeling, cell motility, excitation-contraction coupling, non-associative learning and embryogenesis, just to mention some, through non-ionic functions. When defective, non-ionic functions can give rise to channelopathies involved in cancer, neurodegenerative disease and brain trauma. Ion channels exert their non-ionic functions through a variety of mechanisms that range from physical coupling with other proteins, to possessing enzymatic activity, to assembling with signaling molecules. In this article, we take stock of the field and review recent findings. The concept that emerges, is that one of the most common ways through which channels acquire non-ionic attributes, is by assembling with integrins. These integrin-channel complexes exhibit broad genotypic and phenotypic heterogeneity and reveal a pleiotropic nature, as they appear to be capable of influencing both physiological and pathological processes.
Non-ionic functions of ion channels
Ion channels comprise a fundamental class of integral membrane proteins spanning all three domains of life. Channels make the lipid membrane permeable to ions and for this reason they are present in virtually any cell type [Citation1]. Historically, channels have been considered in light of their conducting properties that range from shaping electrical impulses in excitable cells, to controlling cell volume, secretion, acidification, and other functions in non-excitable cells. However, during the last two decades, we and others, put forward the concept that channels can affect cellular processes in ways that do not depend on their conducting properties (non-ionic functions) [Citation2–9]. Much progress has been achieved, and it is now established that channels possess non-ionic functions that they exert through a number of different mechanisms.
In skeletal muscle fibers, voltage-gated L-type calcium channels CACNA1S (Cav1.1) operate as voltage sensors of excitation-contraction (EC) coupling. The CACNA1S channels are physically, and functionally paired, to the ryanodine receptors type 1 (RyR1, ) [Citation10–12]. Thus, CACNA1Ss promote sarcoplasmic calcium increase through two distinct mechanisms: by conducting a calcium current–the effective contribution of which to EC is controversial (see ref. [Citation13]); and by favoring the release of calcium from the stores through their physical, and thus non-ionic, coupling to the RyR1s. Notably, L-type calcium channels and ryanodine receptors are also functionally coupled in the neurons of the brain. The formation of these macromolecular structures is, in turn, helped by delayed rectifier and voltage-gated potassium channel sub-family 2 member 1 (KCNB1, Kv2.1) that acts as scaffolding agent [Citation14–17]. KCNB1 is expressed in several neuron types in the brain and in other organs including eyes, pancreas, gastrointestinal tract, kidney, and female reproductive system, where it presumably carries an important repolarizing potassium current [Citation18]. However, KCNB1 exhibits a broad range of non-ionic functions, such as acting as scaffold protein as noted above that will be discussed throughout this review.
Figure 1. Ion channels exercise non-ionic functions through multiple mechanisms (a) Channels perform non-ionic functions through physical coupling with other channels, or posses enzymatic domains in their α-subunits (b), or β-subunits (c). In addition to playing canonical regulatory roles, the β-subunits of Nav channels act as adhesion molecules that help forming cell-to-cell contacts, or link the actin cytoskeleton to the extracellular matrix, (d).
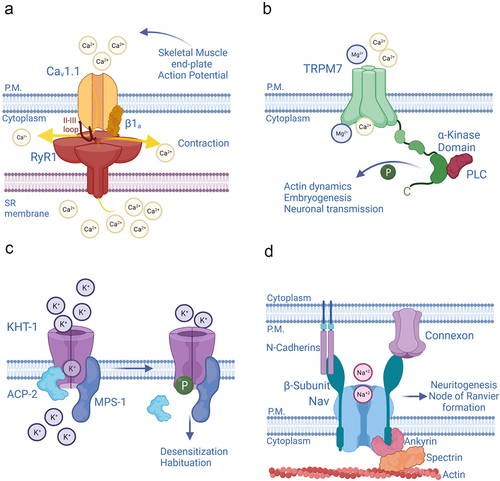
Ion channels have a wide repertoire of mechanisms through which to exert non-ionic functions. For example, the transient receptor potential cation channel melastatin-subfamily, member 7 (TRPM7), a non-selective channel, has enzymatic properties () [Citation3]. The C-terminus of TRPM7 contains a serine/threonine kinase traceable to the α-kinases family [Citation19,Citation20]. TRPM7 can phosphorylate several substrates, including proteins involved in actin dynamics, embryogenesis, and neuronal transmission, underscoring the prominent physiological role of TRPM7 [Citation21–24]. In addition, the α-kinase of TRPM7 indirectly regulates the channel’s ionic properties by modulating its sensitivity to magnesium ions and to magnesium nucleotides [Citation25].
Functional ion channels are seldom the result of a single-gene product. Typically, they are composed of pore-forming or α-subunits, which assemble together to form the pathway for ions (pore) and thus, a conducting channel, and accessory or β-subunits that modulate the properties of the α-subunits. Several accessory subunits of potassium (K+) channels, including mammalian Kvβ2, Drosophila melanogaster slowpoke channel-binding protein SLOB, and Caenorhabditis elegans MiRP K+ channel accessory Subunit (MPS-1) have enzymatic attributes [Citation2,Citation4,Citation5]. MPS-1 is an integral membrane protein, homolog to mammalian KCNEs, that has a cytoplasmic domain capable of serine/threonine kinase activity [Citation26,Citation27]. MPS-1 forms complexes with multiple pore-forming subunits including K+ channel Voltage-Sensitive Subunit 1 (KVS-1) and K+ channel Habituation to Tap subunit 1 (KHT-1) in the nervous system of the worm [Citation9,Citation27]. MPS-1 phosphorylates the α-subunits to decrease their open probability. This mechanism plays an important role in the context of non-associative learning, a universal behavior whereby an organism learns to ignore stimuli that are not important. In the mechanosensory neurons of C. elegans, MPS-1 forms a tripartite complex with KHT-1 and with the acid phosphatase ACP-2, which maintains KHT-1 in a basal, de-phosphorylated state () [Citation9,Citation28]. These tripartite complexes mediate the neurons’ response to mechanical stimuli, such as taps to the Petri dish. When the taps are repeated at constant frequency, the animals habituate. The repetitive stimulus triggers the disengagement of ACP-2 from the complex, allowing MPS-1 to phosphorylate KHT-1. This results in a decrease of K+ efflux that delays touch-neuron repolarization and, as a consequence, produces temporary desensitization to the mechanical stimuli. Notably, in the neurons of the gigantocellular reticular nucleus, the ACP-2 mammalian homolog, prostatic acid phosphatase (PAP) dephosphorylates KHT-1 homolog, murine KCNC1b, where it may probably underlie adaptation responses [Citation28].
Voltage-gated sodium channels (Nav) acquire non-ionic functions through their β-subunits. These proteins not only modulate the ionic properties of the α-subunits; they also mediate cell-to-cell adhesions by acting as molecular linkers that bridge the actin cytoskeleton to neighboring cells. Thus, the β-subunits connect the Nav with the outside, through interacting with proteins such as neurofascins, N-cadherins, and connexins and with the inside, by attaching to the actin cytoskeleton via assembly with ankyrins (. The β-subunit promotes direct bonding of Ankyrin G to the Nav α-subunit) [Citation6,Citation29–32]. This non-ionic function likely contributes to the distribution of Nav channels in zones where they accumulate at high densities such as the node of Ranvier and the hillock, and to neuritogenesis, as some β-subunits (β1) augment, whereas others (β2) inhibit neurite’s outgrowth in cerebellar granule neurons [Citation33].
Integrin-ion channel complexes
A number of ion channels form physical connections with the actin cytoskeleton and with the external environment. However, rather than through their accessory subunits, channels typically achieve those non-ionic functions by interacting with integrins. These are adhesion molecules that connect the extracellular matrix (ECM) to the actin cytoskeleton to regulate the shape, orientation, and movement of cells [Citation34,Citation35]. In addition, integrins engage intracellular signaling pathways, to control cell proliferation (in the absence of integrin-mediated adhesion and growth factors, cells do not commit to enter the cell cycle, [Citation36]), differentiation, survival, and death (anoikis) [Citation37].
Integrin signal transduction is complex and extensively interconnected; therefore, a detailed discussion is beyond the scope of this review (for further readings, we direct the reader to refs. [Citation34,Citation35]). Briefly, in response to anchorage-dependent signals, integrins recruit and/or associate with, the integrin adhesome–a cytoskeletal and signaling complex–to control a multitude of cellular functions [Citation38–40]. Integrins do not possess enzymatic attributes. A fundamental step is the recruitment of Focal Adhesion kinase (FAK), which autophosphorylates Tyr397, creating a binding site for Src kinases. Then, Src, alone or with FAK, phosphorylate several substrates, thereby transducing integrin signals into biochemical events. Integrins promote cell migration by recruiting cytoskeletal linkers, including Talin, and Vinculin, and scaffold/adaptors such as Paxillin and Integrin-linked kinase (ILK) that connect the cytoplasmic tail of integrins to actin filaments. In addition, integrins support cell survival, differentiation, and proliferation, by engaging Ras GTPases, via guanosine-triphosphate exchange factor mSOS. Ras activates several signaling pathways including Mitogen Activated Protein kinase (MAPK) cascades and Phosphoinositide 3 kinase-Protein kinase B-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling that lead to the phosphorylation of cytoplasmic targets and to the enhancement or repression of nuclear transcription.
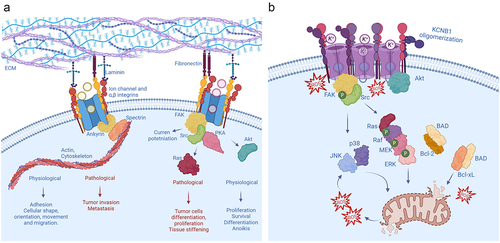
Integrin-channel relationships play prominent roles in disease, primarily cancer, given that several aspects of these pathologies, for instance, tumor invasion, differentiation, and metastasis involve cellular functions that require the coordinated action of integrins (). In addition, the expression of channels or integrins may adjust the expression of one another to maintain cellular homeostasis. In these cases, increased integrin expression may be associated with channel’s recruitment to the plasma membrane and/or boosted channel activity and vice versa. It is also possible that in extreme and/or pathological conditions the increased presence of one may compensate for the absence of the other [Citation41–43]. The first example of cooperation between ion channels and integrins comes from L-type calcium channels expressed in smooth muscle cells of small blood vessels. In rat arterioles, integrin signaling favors vasoconstriction by potentiating CACNA1C (Cav1.2) channels [Citation44–46]. Integrin ligands induce the assembly of integrins α5β1 with CACNA1C channels to form macromolecular complexes with protein kinase A (PKA), and c-Src tyrosine kinases [Citation47]. Following integrin engagement, c-Src phosphorylates CACNA1C leading to current potentiation that facilitates vasoconstriction [Citation48]. Interestingly, α5β1 integrins modulate large conductance, calcium-activated K+ (BK) channels in the same vessels, through largely similar mechanisms [Citation49,Citation50]. The mechanosensitive ion channel PIEZO1 provides an example of a channel regulating integrin signaling [Citation51]. PIEZO1 is overexpressed in aggressive cancers at focal adhesions where it augments tissue stiffening and tumor cell proliferation [Citation52,Citation53]. An increase in PIEZO1 current causes the activation of integrin-FAK signaling that reinforces tissue hardening. In turn, the firmer mechanical microenvironment boosts PIEZO1 expression and promotes tumor cell proliferation in a sort of auto-catalytic process [Citation54]. Another example of a channel able to modulate integrin function is chloride intracellular channel, CLIC1. This protein promotes integrin-mediated, cell-matrix adhesion and the signaling for cytoskeleton extension during tumor cell migration and invasion [Citation55]. Integrins signal through FAK and the critical role of this protein kinase in promoting cell motility is well established [Citation56,Citation57]. In some cancer types, however, FAK activity is decreased and with that, cellular proliferation and invasion. This inhibition of FAK activity is mediated by the formation of tripartite FAK, integrin-β4, and calcium-activated chloride channel protein (mCLCA1) complexes. Thus, overexpression of the same leads to phosphorylation and inhibition of FAK and ERK proteins [Citation58].
Figure 2. Non-ionic functions of integrin channel complexes (a) A widespread mechanism by which channels acquire non-ionic functions is by forming macromolecular complexes with integrins. Generally, these integrin-channel complexes regulate the shape, orientation, and movement of cells through the integrin machinery. In addition, they modulate cell proliferation, differentiation, survival and death, and are therefore implicated in a variety of oncogenic processes of different etiologies. (b) Integrin-α5-KCNB1 complexes operate in the neurons of the brain (for simplicity the ECM is not depicted). Under conditions of oxidative stress, the KCNB1 channels form oligomers that trigger apoptosis. The molecular steps underlying this process include the engagement of FAK and Src by the integrins, followed by the activation of a canonical Ras-MAPK cascade. Killer kinases, such as JNK and p38 and caspases execute the apoptotic program. At the same time, these IKCs neutralize a major mechanism of cell survival by sequestering Akt, that cannot be activated (phosphorylated) and released into the cytoplasm, to phosphorylate its multiple substrates including BAD.
Integrin-K+ channel complexes are widely expressed
Several macromolecular complexes formed by integrins and K+ channels, generically named Integrin-K+ channel complexes or IKCs, have been identified and characterized [Citation41,Citation43,Citation49,Citation57,Citation59–73]. Arcangeli and colleagues were the first to reveal the existence of pathophysiological links between integrins and K+ channels, when they showed that murine erythroleukemia cell adhesion to fibronectin, and neurite outgrowth of neuroblastoma cells were associated with a potassium current, later attributed to the voltage-gated K+ channel KCNH2 (synonyms, Kv11.1, HERG, and hERG1) [Citation67,Citation74–78]. Those initial findings were followed by a series of seminal studies that demonstrated that integrin-mediated cell adhesion of KCNH2 promotes cell differentiation. Most importantly, the activation of KCNH2 channels proceeds through integrin-β1 which was demonstrated to physically interact with the channel [Citation70]. Overall, that body of work shows that integrin-β1-KCNH2 complexes integrate the signaling evoked by cell adhesion to the ECM, with the cell differentiation machinery. The pathological implications of integrin-β1-KCNH2 complexes are significant, when one considers that cell differentiation can play a role in oncogenesis. Accordingly, KCNH2 is highly conserved in tumors of different histogenesis, and integrin-β1-KCNH2 complexes play a central role in cancer formation and progression [Citation72,Citation74,Citation76,Citation79,Citation80]. Furthermore, in colorectal cancer cell lines, the binding of fibronectin and collagen I to integrin-β1 at the ECM level promotes the formation of a tripartite complex composed, other than of integrin-β1, of KCNH2 and of the Na+/H+ antiporter NHE1. The activity of this tripartite complex regulates the cytosolic pH of colorectal cancer cells, thereby contributing to the maintenance of tumor microenvironment [Citation81].
The voltage-gated K+ channel KCNB1 forms stable complexes with integrin-α5 in neurons of the brain [Citation62,Citation64]. It is also likely that these complexes exist in other tissues, including the retina and the pancreas, where both KCNB1 and integrin-α5 are present. Studies carried out in heterologous expression systems indicated that the activity of KCNB1 is translated by the integrins into biochemical events–mediated by FAK, Src, Ras GTPases, MAPKs, and protein kinase Akt–to advance the development of actin-rich cellular protrusions in Chinese hamster ovary (CHO) cells, stimulating their motility and to enhance neuritogenesis of neuroblastoma cells () [Citation57,Citation62–65,Citation82,Citation83]. Accordingly, Src-mediated phosphorylation of KCNB1 at Tyr124 is critical for the proliferation and myelination of murine Schwann cells [Citation84,Citation85]. These functions are predominantly non-ionic in nature. Indeed, while certain non-conducting KCNB1 variants implicated in severe epileptic syndromes engage integrin signaling and stimulate cell migration, other (non-conducting) variants that fail to activate the same cascades do not enhance cell migration [Citation63]. Notably, all those variants are associated with large phenotypic heterogeneity [Citation86]. Overall, it appears that integrin-α5-KCNB1 complexes translate membrane excitability into intracellular signals important for cellular plasticity. This implies that when defective, integrin-α5-KCNB1 complexes could cause neurological disease through a variety of mechanisms ranging from impaired conduction to dysregulated integrin signaling. In fact, the non-ionic, pleiotropic, nature of integrin-α5-KCNB1 complexes becomes relevant in neurodegenerative diseases, when neurons are subject to stressful conditions, namely oxidative stress.
Integrin-α5-KCNB1 complexes trigger programmed cell death
The non-ionic functions of KCNB1 began to unravel after it was discovered that KCNB1 is a pro-apoptotic protein. Aizenman and collaborators showed that cells expressing KCNB1 undergo apoptosis when challenged by oxidants [Citation87]. Subsequent studies revealed that oxidative stress (an imbalance between the oxidants present in the cell and its antioxidant defenses) correlated well with the activation of a number of protein kinases, including p38 MAPK and Src family of tyrosine kinases that phosphorylated KCNB1 at Ser800 and Tyr124 (the latter residue is also responsible for Src-mediated increase of myelination and proliferation of mouse Schwann cells) [Citation84,Citation85,Citation88–91]. The phosphorylation of the channel enhances its interactions with syntaxin accelerating insertion into the plasma membrane [Citation92]. The increased K+ efflux that follows, stimulates caspase and nuclease activity and marks a point of no-return toward apoptosis. However, we later discovered that reactive oxygen species (ROS) can directly modify KCNB1 proteins, turning them into aberrant, toxic channels [Citation93]. The journey into what at the time was uncharted territory began when we tested the idea that the excess ROS that build up in aging cells may oxidize K+ channels, leading to neuronal failure. For that exploratory inquiry, we took advantage of the simplicity of C. elegans, which indeed turned out to be an excellent tool to capture the essence of the problem. Accordingly, the KVS-1 channel, which is a homolog of KCNB1, becomes progressively oxidated at Cys113 in the sensory neurons of aging worms [Citation27,Citation94]. The oxidative modifications alter the gating of KVS-1, which by impairing sensory neuron excitability, leads to behavioral deficit. The cysteine responsible for the functional alterations of the KVS-1 channel is conserved in KCNB1 (Cys73. The KVS-1 channel possesses a 40 amino-acid domain composed of a N-inactivating ball preceded by an N inactivation regulatory domain, called NIRD which modulates the inactivation of the channel [Citation95]). This suggests that also KCNB1 may be susceptible to redox. In fact, oxidants cross-link KCNB1 subunits to each other (oligomers), by inducing the formation of disulfide bridges involving conserved Cys73 [Citation93]. Most importantly, KCNB1 oligomers were detected in the post mortem hippocampi of male and female AD donors (83.8 ± 0.79 yrs. average age) where they were significantly more abundant than in age-matched controls (82.5 ± 0.76 yrs.) [Citation64]. Just to give an idea of the extent of KCNB1 oligomerization in the aging human brain, ~40% of KCNB1 channels were found to be oligomerized in control donors, and this number increased to ~75% in Alzheimer’s donors. Similarly, KCNB1 oligomers were found to be ~35% and ~80% in, respectively, 22 month-old control and 3xTg-AD mice [Citation93]. KCNB1 oligomers do not conduct current [Citation93]. Studies in 3xTg-AD mouse model of Alzheimer’s disease–a pathology characterized by extensive oxidative stress–showed that oxidized KCNB1 channels impair neuron repolarization causing hippocampal hyperexcitability [Citation96,Citation97]. However, the toxicity of KCNB1 oligomers does not stem only from their lack of conduction, and is also caused by non-ionic mechanisms. A KCNB1 variant obtained by replacing Cys73 to Ala (C73A) does not form oligomers and conducts normally. Therefore, C73A channels should give rise to an apoptotic current surge in response to an oxidative insult. In contrast, the mutant does not cause cellular death. This implies that the formation of oligomers, rather than KCNB1 current, is the event that triggers the initial pro-apoptotic stimulus.
Integrin-α5-KCNB1 complexes are implicated in multiple pathologies
Alzheimer’s disease and Traumatic Brain Injury (TBI) provide two well characterized examples of the pleiotropic nature of integrin-α5-KCNB1 complexes. As the brain undergoes degeneration or trauma, these IKCs turn pathogenic by promoting inflammation and apoptosis via integrins and their signaling machinery [Citation64,Citation65,Citation98]. The signaling pathways recruited by integrin-α5-KCNB1 complexes have been characterized in detail. Conformational changes in KCNB1 such as opening and closing leads to the recruitment of FAK. The kinase autophosphorylates Tyr397, creating a binding site for Src family tyrosine kinases. FAK/Src complexes activate small GTPases of the Ras sub-family, which in turn, set in motion a canonical MAPK pathway, composed of Rapidly Activated fibrosarcoma (RAF) kinase, Mitogen-Activated Protein Kinase Kinase (MEK) and Extracellular signal-Regulated kinase (ERK) [Citation62,Citation65]. This is followed by the appearance of killer kinases, including C-Jun N-Terminal Kinase (JNK) and presumably kinases implicated in the surge mechanism such as p38 MAPK, caspases and other death proteins, that execute the final steps of the apoptotic program [Citation82,Citation89,Citation98].
Integrin-α5-KCNB1 complexes keep balance between cell’s life and death
Studies of integrin-α5-KCNB1 complexes expressed in immortal cells demonstrated that the activation of Ras-MAPK signaling represents a causative step toward apoptosis [Citation63,Citation65]. However, also IKCs formed with anti-apoptotic C73A subunits turned out to engage the same Ras-MAPK cascades. The answer to this conundrum is that the difference between IKCs formed with WT and C73A KCNB1 channels resides in the way they regulate active Akt, a major architect of cell survival. Akt keeps apoptosis progression in check through phosphorylating BCL2 associated agonist of cell death (BAD) at Ser136 [Citation99,Citation100]. When BAD is dephosphorylated, it forms heterodimers with Bcl-2 and Bcl-xL, preventing them from inhibiting the release of cytochrome c through the mitochondrial pore [Citation100]. Accordingly, Akt is significantly more active, and with it, BAD is more phosphorylated, in the presence of IKCs formed with C73A mutants compared to WT [Citation63,Citation65]. Furthermore, pharmacological inhibition of Akt abolishes the protective effect of C73A, but when Ras-MAPK signaling is simultaneously inhibited, apoptosis is also suppressed. The activation of Akt occurs at the plasma membrane, where the kinase is sequentially phosphorylated at Thr308 and at Ser473 before being released back into the cytoplasm [Citation101]. An oxidative insult causes selective increase of Akt binding to WT channels that prevents the kinase from being phosphorylated and released into the cytoplasm [Citation65]. At the moment, the causes for the selective affinity of oxidized WT channels for Akt are not known. An important fact to consider, is that KCNB1 oligomers are poorly endocytosed and consequently build up in the plasma membrane [Citation82]. Hence, it is possible that the increased presence of KCNB1 protein at the membrane might enhance the probability of interacting with Akt. Indeed, under normal conditions, a small fraction of KCNB1 channels, either WT and C73A, co-immunoprecipitate with Akt [Citation65]. In summary, the evidence at hand provides a model for the toxicity of integrin-α5-KCNB1 complexes that predicts that these IKCs send apoptotic stimuli via Ras-MAPK cascades, while simultaneously neutralizing the mechanisms of cellular survival.
Biomedical relevance of integrin-α5-KCNB1 complexes
The elucidation of the non-ionic functions of IKCs carries important biomedical implications, as drugs that impinge on the signaling pathways engaged by these complexes have the potential to ameliorate multiple pathologies. One promising candidate is Dasatinib, a second-generation Src tyrosine kinase inhibitor. Dasatinib is FDA-approved for the treatment of Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) and acute lymphoblastic leukemia including Central Nervous System CMS (the drug is blood–brain barrier permeable) [Citation102–110]. Dasatinib, reverses cognitive decline in rodent models of AD by decreasing β-amyloid (Aβ) load and neurofibrillary tau tangles (NFT), inflammation, and oxidative stress [Citation64,Citation111–114]. The potential therapeutic effects of Dasatinib in AD stem from its ability to impinge on multiple cellular mechanisms, which share the involvement of Src tyrosine kinases, such as the oxidation of integrin-α5-KCNB1 complexes [Citation64,Citation98]. Combs and colleagues were the first to show that the inhibition of Src tyrosine kinases by Dasatinib acted to reduce brain inflammation and improved cognitive outcome in mouse model of Alzheimer’s disease [Citation112,Citation113]. Work from our lab further indicated that prolonged Dasatinib treatment in 3xTg-AD mice decreased brain inflammation and Aβ load and reduced behavioral deficit caused by the oxidation of integrin-α5-KCNB1 complexes [Citation64]. Dasatinib was also found to significantly decrease inflammation and neurodegeneration caused by oxidation of integrin-α5-KCNB1 complexes in the Lateral Fluid Percussion (LFP) mouse model of brain trauma, a condition that shares with Alzheimer’s disease copious oxidative stress and Aβ plaque formation [Citation115–120]. Orr and colleagues detected a reduction in total NFT density, neuron loss, and ventricular enlargement following Dasatinib+Quercetin (a flavonoid found in many plants and foods) regimen in human Alzheimer’s neurons and in the brains of a mouse model of tauopathy [Citation111]. Others have sought to identify agents targeting the surge mechanism, which relies on the interaction between the C-terminus of KCNB1 and syntaxin. This effort has led to the identification of a small molecule inhibitor (cpd5) of the protein–protein interaction between syntaxin and KCNB1, that has shown some efficacy in ameliorating neuronal loss in middle cerebral artery occlusion mouse model of ischemic stroke [Citation121]. Currently, seven drugs that inhibit Ras-MAPK signaling are FDA-approved for the treatment of multiple cancer pathologies, and Akt agonists are being developed [Citation122–125]. It is therefore to be hoped that in the future, some of the drugs that target components of the integrin-α5-KCNB1 complexes signaling machinery could be repurposed for the treatment of diseases, including Alzheimer’s disease, TBI, and stroke.
Conclusions
In a short period of time, enormous progress has been achieved in our understanding of ion channels and their non-ionic functions. In 20 years, what started as sporadic, anecdotal evidence has become a solid, broad field of research. Channels exert their non-ionic functions through various mechanisms, ranging from physical coupling, to possessing enzymatic features, and many other mechanisms will likely be discovered as our understanding of these proteins progresses. The repertoire of non-ionic functions of ion channels is implicated in a broad range of physiological processes, as fundamental as actin cytoskeleton remodeling and cell migration, differentiation, embryogenesis, excitation-contraction coupling, and learning and memory formation. Consequently, defective non-ionic functions give rise to pathologies, including TBI, Alzheimer’s disease, stroke, and cancer.
Interestingly, channels achieve their non-ionic functions by primarily interacting with integrins. The voltage-gated and delayed rectifier K+ channel KCNB1 provides one of the best examples of the broad pathophysiological implications of non-ionic functions of an ion channel. Integrin-α5-KCNB1 complexes are involved in regulating basic cellular processes, and they become toxic in pathological conditions. The elucidation of these mechanisms may provide pharmacological indications that could be quickly translated to human clinical trials.
Author contributions
E. Forzisi performed literature search and contributed to the writing of the manuscript. F. Sesti wrote the manuscript
Acknowledgments
We thank Cinzia Sesti for help with the graphics. Figures were created with BioRender.com.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hille B. Ionic channels of excitable membranes. 3rd ed. Sunderland MA: Sinauer Associates; 2001.
- Cai SQ, Hernandez L, Wang Y, et al. MPS-1 is a K(+) channel beta-subunit and a serine/threonine kinase. Nat Neurosci. 2005;8(11):1503–1509.
- Runnels LW, Yue L, Clapham DE. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 2001;291(5506):1043–1047.
- Weng J, Cao Y, Moss N, et al. Modulation of voltage-dependent Shaker family potassium channels by an aldo-keto reductase. J Biol Chem. 2006;281(22):15194–15200.
- Zeng H, Fei H, Levitan IB. The slowpoke channel binding protein Slob from drosophila melanogaster exhibits regulatable protein kinase activity. Neurosci Lett. 2004;365(1):33–38.
- Malhotra JD, Kazen-Gillespie K, Hortsch M, et al. Sodium channel β subunits mediate homophilic cell adhesion and recruit ankyrin to Points of cell-cell contact *. J Biol Chem. 2000;275(15):11383–11388.
- Mochida S, Yokoyama CT, Kim DK, et al. Evidence for a voltage-dependent enhancement of neurotransmitter release mediated via the synaptic protein interaction site of N-type Ca 2+ channels. Proc Natl Acad Sci U S A. 1998;95(24):14523–14528.
- Dolmetsch RE, Pajvani U, Fife K, et al. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294(5541):333–339.
- Cai SQ, Wang Y, Park KH, et al. Auto-phosphorylation of a voltage-gated K+ channel controls non-associative learning. EMBO J. 2009;28(11):1601–1611.
- Inui M, Saito A, Fleischer S. Purification of the ryanodine receptor and identity with feet structures of junctional terminal cisternae of sarcoplasmic reticulum from fast skeletal muscle. J Biol Chem. 1987;262(4):1740–1747.
- Eisenberg RS, McCarthy RT, Milton RL. Paralysis of frog skeletal muscle fibres by the calcium antagonist D-600. J Physiol. 1983;341(1):495–505.
- Tanabe T, Beam KG, Powell JA, et al. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 1988;336(6195):134–139.
- Flucher BE. Skeletal muscle CaV1.1 channelopathies. Pflugers Arch. 2020;472(7):739–754.
- Vierra NC, Kirmiz M, van der List D, et al. Kv2.1 mediates spatial and functional coupling of L-type calcium channels and ryanodine receptors in mammalian neurons. Elife. 2019;8:e49953.
- Kirmiz M, Palacio S, Thapa P, et al. Remodeling neuronal ER-PM junctions is a conserved nonconducting function of Kv2 plasma membrane ion channels. Mol Biol Cell. 2018;29(20):2410–2432.
- Fox PD, Haberkorn CJ, Akin EJ, et al. Induction of stable ER-plasma-membrane junctions by Kv2.1 potassium channels. J Cell Sci. 2015;128(11):2096–2105.
- Johnson B, Leek AN, Solé L, et al. Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc Natl Acad Sci U S A. 2018;115(31):E7331–40.
- Sesti F, Wu X, Liu S. Oxidation of KCNB1 K(+) channels in central nervous system and beyond. World J Biol Chem. 2014;5(2):85–92.
- Ryazanov AG, Ward MD, Mendola CE, et al. Identification of a new class of protein kinases represented by eukaryotic elongation factor-2 kinase. Proc Natl Acad Sci U S A. 1997;94(10):4884–4889.
- Ryazanova LV, Dorovkov MV, Ansari A, et al. Characterization of the protein kinase activity of TRPM7/ChaK1, a protein kinase fused to the transient receptor potential ion channel. J Biol Chem. 2004;279(5):3708–3716.
- Jin J, Desai BN, Navarro B, et al. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg 2+ homeostasis. Science. 2008;322(5902):756–760.
- Krapivinsky G, Mochida S, Krapivinsky L, et al. The TRPM7 ion channel functions in cholinergic synaptic vesicles and affects transmitter release. Neuron. 2006;52(3):485–496.
- Clark K, Langeslag M, van Leeuwen B, et al. TRPM7, a novel regulator of actomyosin contractility and cell adhesion. EMBO J. 2006;25(2):290–301.
- Krapivinsky G, Krapivinsky L, Manasian Y, et al. The TRPM7 chanzyme is cleaved to release a chromatin-modifying kinase. Cell. 2014;157(5):1061–1072.
- Demeuse P, Penner R, Fleig A. TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J Gen Physiol. 2006;127(4):421–434.
- McCrossan ZA, Abbott GW. The MinK-related peptides. Neuropharmacology. 2004;47(6):787–821.
- Bianchi L, Kwok SM, Driscoll M, et al. A potassium channel-MiRP complex controls neurosensory function in caenorhabditis elegans. J Biol Chem. 2003;278(14):12415–12424.
- Cotella D, Hernandez-Enriquez B, Duan Z, et al. An evolutionarily conserved mode of modulation of Shaw -like K + channels. FASEB J. 2013;27(4):1381–1393.
- McEwen DP, Isom LL. Heterophilic interactions of sodium channel beta1 subunits with axonal and glial cell adhesion molecules. J Biol Chem. 2004;279(50):52744–52752.
- Ratcliffe CF, Westenbroek RE, Curtis R, et al. Sodium channel beta1 and beta3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol. 2001;154(2):427–434.
- McEwen DP, Meadows LS, Chen C, et al. Sodium channel beta1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin. J Biol Chem. 2004;279(16):16044–16049.
- Pan Z, Kao T, Horvath Z, et al. A Common Ankyrin-G-based mechanism retains KCNQ and Na V channels at electrically active domains of the axon. J Neurosci. 2006;26(10):2599–2613.
- Davis TH, Chen C, Isom LL. Sodium channel beta1 subunits promote neurite outgrowth in cerebellar granule neurons. J Biol Chem. 2004;279(49):51424–51432.
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285(5430):1028–1032.
- Cooper J, Giancotti FG. Integrin signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. 2019;35(3):347–367.
- Walker JL, Assoian RK. Integrin-dependent signal transduction regulating cyclin D1 expression and G1 phase cell cycle progression. Cancer Metastasis Rev. 2005;24(3):383–393.
- Kim C, Ye F, Ginsberg MH. Regulation of integrin activation. Annu Rev Cell Dev Biol. 2011;27(1):321–345.
- Winograd-Katz SE, Fässler R, Geiger B, et al. The integrin adhesome: from genes and proteins to human disease. Nat Rev Mol Cell Biol. 2014;15(4):273–288.
- Horton ER, Byron A, Askari JA, et al. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol. 2015;17(12):1577–1587.
- Zaidel-Bar R, Itzkovitz S, Ma’ayan A, et al. Functional atlas of the integrin adhesome. Nat Cell Biol. 2007;9(8):858–867.
- Song MS, Choi SY, Ryu PD, et al. Voltage-Gated K+ channel, Kv3.3 is involved in hemin-induced K562 differentiation. PLoS One. 2016;11(2):e0148633.
- Atcha H, Meli VS, and Davis CT, et al. Crosstalk between CD11b and Piezo1 mediates macrophage responses to mechanical cues. Front Immunol. 2021;12.
- Wright JR, Jones S, Parvathy S, et al. The voltage-gated K(+) channel Kv1.3 modulates platelet motility and α(2)β(1) integrin-dependent adhesion to collagen. Platelets. 2022;33:451–461.
- Wu X, Mogford JE, Platts SH, et al. Modulation of calcium current in arteriolar smooth muscle by alphav beta3 and alpha5 beta1 integrin ligands. J Cell Biol. 1998;143(1):241–252.
- Wu X, Davis GE, Meininger GA, et al. Regulation of the L-type calcium channel by alpha 5 beta 1 integrin requires signaling between focal adhesion proteins. J Biol Chem. 2001;276(32):30285–30292.
- Waitkus-Edwards KR, Martinez-Lemus LA, Wu X, et al. α 4 β 1 integrin activation of L-type calcium channels in vascular smooth muscle causes arteriole vasoconstriction. Circ Res. 2002;90(4):473–480.
- Chao J-T, Gui P, Zamponi GW, et al. Spatial association of the Cav1.2 calcium channel with α 5 β 1 -integrin. Am J Physiol Cell Physiol. 2011;300(3):C477–489.
- Gui P, Wu X, Ling S, et al. Integrin receptor activation triggers converging regulation of Cav1.2 calcium channels by c-Src and protein kinase A pathways. J Biol Chem. 2006;281(20):14015–14025.
- Yang Y, Wu X, Gui P, et al. Alpha5beta1 integrin engagement increases large conductance, Ca2+-activated K+ channel current and Ca2+ sensitivity through c-src-mediated channel phosphorylation. J Biol Chem. 2010;285(1):131–141.
- Wu X, Yang Y, Gui P, et al. Potentiation of large conductance, Ca 2+ -activated K + (BK) channels by α5β1 integrin activation in arteriolar smooth muscle. J Physiol. 2008;586(6):1699–1713.
- Coste B, Mathur J, Schmidt M, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330(6000):55–60.
- Zhou W, Liu X, van Wijnbergen Jwm, et al. Identification of PIEZO1 as a potential prognostic marker in gliomas. Sci Rep. 2020;10:16121.
- Jetta D, Bahrani Fard MR, Sachs F, et al. Adherent cell remodeling on micropatterns is modulated by Piezo1 channels. Sci Rep. 2021;11(1):5088.
- Chen X, Wanggou S, Bodalia A, et al. A feedforward mechanism mediated by mechanosensitive ion channel PIEZO1 and tissue mechanics promotes glioma aggression. Neuron. 2018;100(4):799–815.e7.
- Peng J-M, Lin S-H, Yu M-C, et al. CLIC1 recruits PIP5K1A/C to induce cell-matrix adhesions for tumor metastasis. J Clin Invest. 2021;131:133525.
- Cáceres M, Ortiz L, Recabarren T, et al. TRPM4 Is a novel component of the adhesome required for focal adhesion disassembly, migration and contractility. PLoS One. 2015;10(6):e0130540.
- Wei J-F, Wei L, Zhou X, et al. Formation of Kv2.1-FAK complex as a mechanism of FAK activation, cell polarization and enhanced motility. J Cell Physiol. 2008;217(2):544–557.
- Qiang -Y-Y, Li C-Z, Sun R, et al. Along with its favorable prognostic role, CLCA2 inhibits growth and metastasis of nasopharyngeal carcinoma cells via inhibition of FAK/ERK signaling. J Exp Clin Cancer Res. 2018;37(1):34.
- Sengupta S, Rothenberg KE, Li H, et al. Altering integrin engagement regulates membrane localization of K(ir)2.1 channels. J Cell Sci. 2019;132(17). DOI:10.1242/jcs.225383
- Birkner K, Wasser B, Ruck T, et al. β1-Integrin- and KV1.3 channel-dependent signaling stimulates glutamate release from Th17 cells. J Clin Invest. 2020;130(2):715–732.
- Girault A, Chebli J, Privé A, et al. Complementary roles of KCa3.1 channels and β1-integrin during alveolar epithelial repair. Respir Res. 2015;16:100.
- Yu W, Gowda M, Sharad Y, et al. Oxidation of KCNB1 potassium channels triggers apoptotic integrin signaling in the brain. Cell Death Dis. 2017;8:e2737.
- Yu W, Shin MR, Sesti F. Complexes formed with integrin-α5 and KCNB1 potassium channel wild type or epilepsy-susceptibility variants modulate cellular plasticity via Ras and Akt signaling. FASEB J. 2019;33(12):14680–14689.
- Wei Y, Shin MR, Sesti F. Oxidation of KCNB1 channels in the human brain and in mouse model of Alzheimer’s disease. Cell Death Dis. 2018;9(8):820.
- Forzisi E, Yu W, Rajwade P, et al. Antagonistic roles of Ras-MAPK and Akt signaling in integrin-K + channel complex-mediated cellular apoptosis. FASEB J. 2022;36(5):e22292.
- Bianchi L, Arcangeli A, Bartolini P, et al. An inward rectifier K+ current modulates in neuroblastoma cells the tyrosine phosphorylation of the pp125FAK and associated proteins: role in neuritogenesis. Biochem Biophys Res Commun. 1995;210(3):823–829.
- Arcangeli A, Becchetti A, Mannini A, et al. Integrin-mediated neurite outgrowth in neuroblastoma cells depends on the activation of potassium channels. J Cell Biol. 1993;122(5):1131–1143.
- Becchetti A, Arcangeli A, Del Bene MR, et al. Response to fibronectin-integrin interaction in leukaemia cells: delayed enhancing of a K+ current. Proc Biol Sci. 1992;248:235–240.
- Cherubini A, Pillozzi S, Hofmann G, et al. HERG K + channels and β1 integrins interact through the assembly of a macromolecular complex. Ann N Y Acad Sci. 2002;973(1):559–561.
- Cherubini A, Hofmann G, Pillozzi S, et al. Human ether-a-go-go -related Gene 1 channels are physically linked to β 1 integrins and modulate adhesion-dependent signaling. Mol Biol Cell. 2005;16(6):2972–2983.
- Hofmann G, Bernabei PA, Crociani O, et al. HERG K+ channels activation during beta(1) integrin-mediated adhesion to fibronectin induces an up-regulation of alpha(v)beta(3) integrin in the preosteoclastic leukemia cell line FLG 29.1. J Biol Chem. 2001;276:4923–4931.
- Duranti C, Iorio J, Lottini T, et al. Harnessing the hERG1/β1 integrin complex via a novel bispecific single-chain antibody: an effective strategy against solid cancers. Mol Cancer Ther. 2021;20(8):1338–1349.
- Levite M, Cahalon L, Peretz A, et al. Extracellular K(+) and opening of voltage-gated potassium channels activate T cell integrin function: physical and functional association between Kv1.3 channels and beta1 integrins. J Exp Med. 2000;191(7):1167–1176.
- Arcangeli A, Bianchi L, Becchetti A, et al. A novel inward-rectifying K+ current with a cell-cycle dependence governs the resting potential of mammalian neuroblastoma cells. J Physiol. 1995;489(Pt 2):455–471.
- Arcangeli A, Faravelli L, Bianchi L, et al. Soluble or bound laminin elicit in human neuroblastoma cells short- or long-term potentiation of a K+ inwardly rectifying current: relevance to neuritogenesis. Cell Adhes Commun. 1996;4:369–385.
- Bianchi L, Wible B, Arcangeli A, et al. herg encodes a K+ current highly conserved in tumors of different histogenesis: a selective advantage for cancer cells? Cancer Res. 1998;58(4):815–822.
- Arcangeli A, Rosati B, Cherubini A, et al. Long-term exposure to retinoic acid induces the expression of IRK1 channels in HERG channel-endowed neuroblastoma cells. Biochem Biophys Res Commun. 1998;244(3):706–711.
- Arcangeli A, Rosati B, Crociani O, et al. Modulation of HERG current and herg gene expression during retinoic acid treatment of human neuroblastoma cells: potentiating effects of BDNF. J Neurobiol. 1999;40(2):214–225.
- Pillozzi S, Brizzi MF, Balzi M, et al. HERG potassium channels are constitutively expressed in primary human acute myeloid leukemias and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia. 2002;16(9):1791–1798.
- Crociani O, Zanieri F, Pillozzi S, et al. hERG1 channels modulate integrin signaling to trigger angiogenesis and tumor progression in colorectal cancer. Sci Rep. 2013;3(1):3308.
- Iorio J, Duranti C, Lottini T, et al. K(V)11.1 potassium channel and the Na(+)/H(+) Antiporter NHE1 modulate adhesion-dependent intracellular pH in colorectal cancer cells. Front Pharmacol. 2020;11:848.
- Wu X, Hernandez-Enriquez B, Banas M, et al. Molecular mechanisms underlying the apoptotic effect of KCNB1 K+ channel oxidation. J Biol Chem. 2013;288(6):4128–4134.
- Hu X, Wei L, Taylor TM, et al. Hypoxic preconditioning enhances bone marrow mesenchymal stem cell migration via Kv2.1 channel and FAK activation. Am J Physiol Cell Physiol. 2011;301(2):C362–72.
- Sobko A, Peretz A, Attali B. Constitutive activation of delayed-rectifier potassium channels by a src family tyrosine kinase in Schwann cells. EMBO J. 1998;17(16):4723–4734.
- Peretz A, Sobko A, Attali B. Tyrosine kinases modulate K + channel gating in mouse Schwann cells. J Physiol. 1999;519(2):373–384.
- de Kovel CGF, Syrbe S, Brilstra EH, et al. Neurodevelopmental disorders caused by de novo variants in KCNB1 genotypes and phenotypes. JAMA Neurol. 2017;74(10):1228–1236.
- Pal S, Hartnett KA, Nerbonne JM, et al. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci. 2003;23(12):4798–4802.
- Aras MA, Aizenman E. Obligatory role of ASK1 in the apoptotic surge of K+ currents. Neurosci Lett. 2005;387(3):136–140.
- Redman PT, He K, Hartnett KA, et al. Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc Natl Acad Sci U S A. 2007;104(9):3568–3573.
- Redman PT, Hartnett KA, Aras MA, et al. Regulation of apoptotic potassium currents by coordinated zinc-dependent signalling. J Physiol. 2009;587(18):4393–4404.
- Tiran Z, Peretz A, Attali B, et al. Phosphorylation-dependent regulation of Kv2.1 channel activity at tyrosine 124 by Src and by protein-tyrosine phosphatase epsilon. J Biol Chem. 2003;278:17509–17514.
- McCord MC, Aizenman E. Convergent Ca 2+ and Zn 2+ signaling regulates apoptotic Kv2.1 K + currents. Proc Natl Acad Sci U S A. 2013;110(34):13988–13993.
- Cotella D, Hernandez-Enriquez B, Wu X, et al. Toxic role of K+ channel oxidation in mammalian brain. J Neurosci. 2012;32:4133–4144.
- Cai SQ, Sesti F. Oxidation of a potassium channel causes progressive sensory function loss during aging. Nat Neurosci. 2009;12:611–617.
- Cai SQ, Sesti F. A new mode of regulation of N-type inactivation in a Caenorhabditis elegans voltage-gated potassium channel. J Biol Chem. 2007;282(25):18597–18601.
- Frazzini V, Guarnieri S, Bomba M, et al. Altered Kv2.1 functioning promotes increased excitability in hippocampal neurons of an Alzheimer’s disease mouse model. Cell Death Dis. 2016;7(2):e2100.
- Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–421.
- Yu W, Parakramaweera R, Teng S, et al. Oxidation of KCNB1 potassium channels causes neurotoxicity and cognitive impairment in a mouse model of traumatic brain injury. J Neurosci. 2016;36(43):11084–11096.
- Bergmann A. Survival signaling goes BAD. Dev Cell. 2002;3(5):607–608.
- Burlacu A. Regulation of apoptosis by Bcl-2 family proteins. J Cell Mol Med. 2003;7(3):249–257.
- Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4(9):a011189.
- Abdelhalim A, Barcos M, Block AW, et al. Remission of Philadelphia chromosome-positive central nervous system leukemia after dasatinib therapy. Leuk Lymphoma. 2007;48(5):1053–1056.
- Porkka K, Koskenvesa P, Lundan T, et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood. 2008;112(4):1005–1012.
- Alimena G, Breccia M, Latagliata R, et al. Dasatinib in the management of lymphoid blast crisis of Philadelphia-positive chronic myeloid leukemia with multiple extra-medullary and intracranial localizations. Leuk Res. 2009;33(8):e134–6.
- Bhadri VA, Satharasinghe K, Sugo E, et al. Excellent response to dasatinib of childhood Philadelphia positive intracranial acute lymphoblastic leukaemia tumours. Br J Haematol. 2011;152(3):347–349.
- Russwurm G, Heinsch M, Radkowski R, et al. Dasatinib induces complete remission in a patient with primary cerebral involvement of B-cell chronic lymphocytic leukemia failing chemotherapy. Blood. 2010;116(14):2617–2618.
- Zhou HS, Dai M, Wei Y, et al. Isolated central nervous system relapse in patient with blast-crisis chronic myeloid leukemia in durable complete cytogenetic remission on dasatinib treatment: pharmacokinetics and ABL mutation analysis in cerebrospinal fluid. Leuk Lymphoma. 2013;54(7):1557–1559.
- Nishimoto M, Nakamae H, Koh KR, et al. Dasatinib maintenance therapy after allogeneic hematopoietic stem cell transplantation for an isolated central nervous system blast crisis in chronic myelogenous leukemia. Acta Haematol. 2013;130(2):111–114.
- Xu Z, Zheng M, Wu C, et al. The overwhelmingly positive response to Dasatinib of a patient with multiple blast crisis of chronic myeloid leukemia. Int J Clin Exp Med. 2015;8(1):1460–1466.
- Lai SW, Huang TC, Chen JH, et al. Dasatinib as the salvage therapy for chronic myeloid leukemia with blast crisis and central nervous system involvement: a case report. Oncol Lett. 2015;9(4):1957–1961.
- Musi N, Valentine JM, Sickora KR, et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 2018;17(6):e12840.
- Dhawan G, Combs CK. Inhibition of Src kinase activity attenuates amyloid associated microgliosis in a murine model of Alzheimer’s disease. J Neuroinflammation. 2012;9(1):117.
- Dhawan G, Floden AM, Combs CK. Amyloid-beta oligomers stimulate microglia through a tyrosine kinase dependent mechanism. Neurobiol Aging. 2012;33(10):2247–2261.
- Zhang P, Kishimoto Y, Grammatikakis I, et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. 2019;22(5):719–728.
- Gentleman SM, Greenberg BD, Savage MJ, et al. A beta 42 is the predominant form of amyloid beta-protein in the brains of short-term survivors of head injury. Neuroreport. 1997;8(6):1519–1522.
- Roberts GW, Gentleman SM, Lynch A, et al. beta A4 amyloid protein deposition in brain after head trauma. Lancet. 1991;338(8780):1422–1423.
- Roberts GW, Gentleman SM, Lynch A, et al. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1994;57(4):419–425.
- Graham DI, Gentleman SM, Lynch A, et al. Distribution of beta-amyloid protein in the brain following severe head injury. Neuropathol Appl Neurobiol. 1995;21(1):27–34.
- Ikonomovic MD, Uryu K, Abrahamson EE, et al. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190(1):192–203.
- Rodríguez-Rodríguez A, Egea-Guerrero JJ, Murillo-Cabezas F, et al. Oxidative stress in traumatic brain injury. Curr Med Chem. 2014;21(10):1201–1211.
- Yeh CY, Bulas AM, Moutal A, et al. Targeting a potassium channel/syntaxin interaction ameliorates cell death in ischemic stroke. J Neurosci. 2017;37(23):5648–5658.
- Cheng Y, Tian H. Current development status of MEK inhibitors. Molecules. 2017;22(10):1551.
- Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer. 2017;17(11):676–691.
- Bu X, Yin C, Zhang X, et al. LaSota strain expressing the rabies virus glycoprotein (rL-RVG) suppresses gastric cancer by inhibiting the alpha 7 nicotinic acetylcholine receptor (α7 nAChR)/Phosphoinositide 3-Kinase (PI3K)/AKT pathway. Med Sci Monit. 2019;25:5482–5492.
- Molina-Arcas M, Samani A, Downward J. Drugging the undruggable: advances on RAS targeting in cancer. Genes (Basel). 2021;12(6):899.