
Abstract
Antagonistic antibodies targeting the G-protein C-X-C chemokine receptor 4 (CXCR4) hold promising therapeutic potential in various diseases. We report for the first time the detailed mechanism of action at a molecular level of a potent anti-CXCR4 antagonistic antibody (MEDI3185). We characterized the MEDI3185 paratope using alanine scanning on all 6 complementary-determining regions (CDRs). We also mapped its epitope using CXCR4 mutagenesis to assess the relative importance of the CXCR4 N-terminal peptide, extracellular loops (ECL) and ligand-binding pocket. We show that the interaction between MEDI3185 and CXCR4 is mediated mostly by CDR3H in MEDI3185 and ECL2 in CXCR4. The MEDI3185 epitope comprises the entire ECL2 sequence, lacks any so-called ‘hot-spot’ and is remarkably resistant to mutations. The structure of MEDI3185 variable domains was modeled, and suggested a β-strand/β-strand interaction between MEDI3185 CDR3H and CXCR4 ECL2, resulting in direct steric hindrance with CXCR4 ligand SDF-1. These findings may have important implications for designing antibody therapies against CXCR4.
Introduction
G-protein C-X-C chemokine receptor 4 (CXCR4) is a 7-transmembrane spanning protein that binds the chemokine stromal derived factor-1 (SDF-1).Citation1-3 This pathway regulates many physiological processes, such as leukocyte trafficking, stem cell mobilization, and embryonic development of the cardiovascular, haematopoietic , and central nervous systems.Citation4-7 Furthermore, CXCR4 plays an important role in many human pathologies, including human immunodeficiency virus (HIV) infection, rheumatoid arthritis, and cancer metastasis and development. In particular, HIV-1 strains use CXCR4 as a co-receptor for viral entry into host cells.Citation8,9 In rheumatoid arthritis, the CXCR4/SDF-1 pathway stimulates the migration of memory T cells and inhibits T cell apoptosis.Citation10 Moreover, CXCR4 is broadly expressed in various types of cancers.Citation11-16 These observations suggest that CXCR4 is an important target to prosecute. To this effect, various inhibitors have been developed including small molecules, peptides and antibodies.Citation17-21 A small molecule antagonist, AMD3100, has shown therapeutic potential for HIV infection, inflammatory diseases, stem-cell mobilization and cancers.Citation22-31 Peptide-based inhibitors such as T22,Citation32 T140Citation33 and their derivativesCitation34,35 have also shown promising activity in various disease models.Citation17
Much efforts have been devoted to characterize the mechanism of action of such inhibitors, including structure-activity relationship studies, computational molecular modeling, and structural characterization.Citation36-41 Recently, crystal structures of CXCR4 bound to a small molecule antagonist (IT1t) and a cyclic peptide (CVX15)Citation40 were reported. Both inhibitors bind into a pocket formed by transmembrane helices I, II, III, and VII. While IT1t occupies only part of this pocket, CVX15 fills most of the volume and also interacts with residues of the second extracellular loop (ECL2). This pocket has been hypothesized to be the signaling trigger for SDF-1 and several acidic residues were identified as crucial for the interaction with SDF-1 or HIV-1.Citation42 Such studies have provided a molecular basis for the antagonistic activity of low-molecular weight CXCR4 inhibitors.
Alternate CXCR4 blocking strategies using mAbs have also been pursued. In particular, both mouse and rat mAbs directed against CXCR4 have shown activity in inhibiting HIV infection and cancer migration/growth in animal models.Citation43-47 More recently, a fully human mAb (BMS-936564) has demonstrated tumor growth inhibition activity and is currently in clinical trials to treat relapsed/refractory hematologic malignancies.Citation20 We have also reported a fully human antagonistic anti-CXCR4 mAb, MEDI3185 (IgG1, κ).Citation21 MEDI3185 can inhibit tumor growth in hematologic tumors as a single-agent and shows combination activity in ovarian tumor models. While interest in developing anti-CXCR4 mAb therapy increases, so does understanding the molecular basis for the corresponding mechanism(s) of action. Several studies have probed the extracellular portion of CXCR4 using mutagenesis, and concluded that anti-CXCR4 mAbs can recognize different portions, such as the N-terminal peptide, ECL1, 2 or 3.Citation43,48-50 However, the molecular mechanisms by which anti-CXCR4 antagonistic mAbs exert their inhibitory activity remain largely unknown.
In an effort to elucidate molecular mechanisms by which mAbs can inhibit CXCR4, we have determined the binding mode of MEDI3185. We characterized the paratope of MEDI3185 using alanine scanning on its 6 complementarity-determining regions (CDRs), mapped its epitope by CXCR4 mutagenesis and modeled the structure of its variable domains. We propose a novel mode of CXCR4 inhibition, different from that of small-molecule/peptide inhibitors.
Results
Determination of MEDI3185 paratope
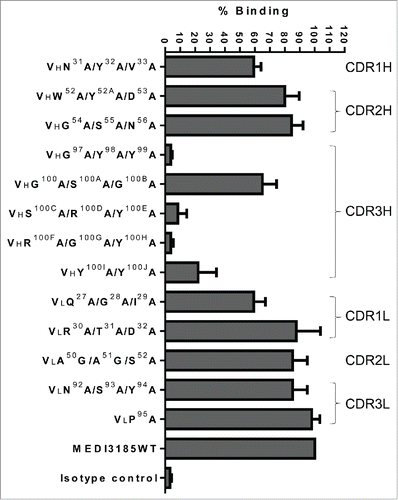
Alanine scanning was performed across all 6 MEDI3185 CDRs to identify its paratope. The variable domain sequences of MEDI3185 are shown in , with CDRs defined as per Kabat.Citation51 Of note, CDR3H comprises 20 amino acids, a relatively long span when compared with the corresponding average length in human (namely 13.1 residues).Citation52 CDR1H, 2H, 1L, 2L and 3L all belong to known canonical classes based on their primary sequences (corresponding to PDB ID numbers 2fbj, 1igc, 1ikf, 1lmk and 1tet, respectively). Thus, we used their corresponding canonical structuresCitation53-55 to select residues at or near the apex of each loop for mutagenesis (excluding positions known to be part of the VL/VH interface)Citation56 because these are more likely to be solvent exposed and antigen-accessible. For CDR3H, a large (15/20) portion of residues in its middle section was chosen for substitutions. Thirteen ‘Ala’ or ‘Gly’ variants, single or in clusters, were then constructed at the following positions (Kabat numbering)Citation51: N31Y32V33 (CDR1H), W52Y52AD53 (CDR2H), G54S55N56 (CDR2H), G97Y98Y99 (CDR3H), G100S100AG100B (CDR3H), S100CR100DY100E (CDR3H), R100FG100GY100H (CDR3H), Y100IY100J (CDR3H), Q27G28I29 (CDR1L), R30T31D32 (CDR1L), A50A51S52 (CDR2L), N92S93Y94 (CDR3L), and P95 (CDR3L).
Figure 1. Amino acid sequence of MEDI3185 VH and VL domains. CDRs (Kabat definition).Citation51 are bold and underlined, whereas residues marked with asterisks indicate positions where mutations were introduced.

MEDI3185 variants were expressed in Chinese hamster ovary (CHO) cells and their binding to human CXCR4 assessed using flow cytometry (). Five variants bearing mutations in CDR2H, 2L or 3L bound similarly well to CXCR4 compared to un-mutated MEDI3185. Mutations in CDR1H (VHN31A/Y32A/V33A) and CDR1L (VLQ27A/G28A/I29A) exhibited slightly decreased binding compared with un-mutated MEDI3185, suggesting some contribution of the corresponding CDRs to the interaction with CXCR4. CDR3H was found to be critical, as 4 out of 5 variants in this loop exhibited substantially decreased or abolished binding to CXCR4 (S100CA/R100DA/Y100EA , Y100IA/Y100JA, G97A/Y98A/Y99A and R100FA/G100GA/Y100H. Therefore, the MEDI3185 paratope mostly comprises CDR3H.
Figure 2. Binding characterization of MEDI3185 variants to CXCR4. Thirteen variants, single or combinatorial, were generated by replacing select CDR residues with Ala or Gly (for A50 and A51 in CDR2L). Binding of MEDI3185 variants was calculated as % binding when compared with wild-type (WT) MEDI3185. Results represent the means of 3 independent experiments.
Determination of MEDI3185 epitope
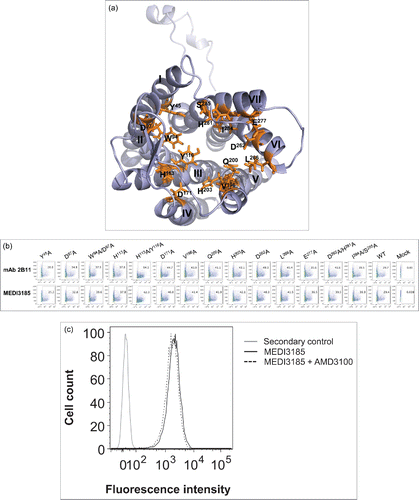
MEDI3185 epitope was identified by mutagenesis of potential solvent-accessible regions on human CXCR4.Citation40 These included transmembrane helices residues defining the ligand-binding pocket,Citation40 the N-terminal peptide and the 3 ECLs. Ala mutations in helices were carried out alone or in combination and included residues Y45, D97, W94, H113, Y116, D171, V196, Q200, H203, L266, D263, E277, H281, I284 and S285 (). All mutants expressed well on the surface of CHO cells () as monitored using mAb 2B11, which recognizes CXCR4 N-terminal peptide.Citation57 MEDI3185 binding to these CXCR4 variants was assessed by flow cytometry. All variants exhibited similar binding compared to wild-type CXCR4 (), suggesting that the ligand binding pocket, although constituting the binding site of small molecule and peptide-based CXCR4 inhibitors, Citation40,58 is not involved in the interaction with MEDI3185. Indeed, the CXCR4 small molecule inhibitor AMD3100 did not affect binding of MEDI3185 to CXCR4 (). Thus, MEDI3185 interacts with CXCR4 with a distinct mode of action.
Figure 3. (a) Three-dimensional representation of human CXCR4 (PDB ID number 3ODU).Citation40 Residues in transmembrane helices whose side chains contribute to the ligand-binding pocket are shown in orange sticks. (b) Binding of MEDI3185 to ligand-binding pocket CXCR4 variants by FACS. CXCR4 expression was monitored using mAb 2B11. The y axis represents side scatter characteristics, while the x axis represents the mean fluorescence intensity (MFI). (c) Competition binding between MEDI3185 and AMD3100. Binding of MEDI3185 to Jurkat cells was not affected in the presence of 10 μM AMD3100.
To probe CXCR4 N-terminal peptide and its 3 ECLs, a series of chimeric human/mouse variants were constructed. More precisely, we generated 8 loss-of-function (knock-out, ‘KO’) variants by replacing human segments with their mouse counterparts, and 2 gain-of-function (knock-in, ‘KI’) by grafting human regions into the mouse molecule (). Murine CXCR4 was selected for generating the chimeric variants because it shares ˜90% sequence identity with human CXCR4 (), and is only faintly recognized by MEDI3185 (). All chimeric variants expressed well on the surface of CHO cells as monitored with anti-CXCR4 mAb 2B11, which recognizes both human and mouse CXCR4 (). The N-terminal peptide, ECL1 and ECL3 did not appear to play a significant role (; KO_Nterm, KO_ECL1, KO_ECL3 and KO_Nterm+ECL3). Only upon substituting human ECL2 with the corresponding mouse residues was MEDI3185 binding reduced to a level comparable to that of mouse CXCR4 (; KO_ECL2, KO_Nterm+ECL2, and KO_ECL2+ECL3). Therefore, MEDI3185 epitope is localized within CXCR4 ECL2. To refine this observation, KO and KI chimeric variants that targeted smaller sections within ECL2 were generated. ECL2 is the largest extracellular loop of CXCR4, and is anchored on helix III via a disulfide bond between Cys186 in ECL2 and Cys109 in helix III.Citation40 It comprises 2 portions, a β-hairpin structure (A175-I185) and a loop on its base (D187-V196). Therefore, 2 KO and 2 KI variants were constructed by swapping each region between human and mouse (). Interestingly, MEDI3185 still bound well to both KO variants (). Likewise, when grafted onto the murine sequence, either portion of human ECL2 conferred good binding to MEDI3185 (). Thus, both ECL2 portions significantly contribute to the interaction with MEDI3185.
Figure 4. (a) Amino acid alignment of human (hu) and mouse (mo) CXCR4. Secondary structural elements are shown according to the crystal structure of CXCR4.Citation40 Underlined sequences were swapped between human and mouse CXCR4 to construct chimeric variants. (b) Nomenclature and schematic representation of KO and KI CXCR4 variants based on the crystal structure of CXCR4.Citation40 The N-terminal peptide was added using Discovery Studio. Ten chimeric variants were constructed by swapping in or out various segments of human (green) into mouse (cyan) CXCR4 (KI), or of mouse into human CXCR4 (KO). (c) MEDI3185 binding to various CXCR4 chimeric variants by FACS. CXCR4 expression was monitored using mAb 2B11. The y axis represents side scatter characteristics, while the x axis represents the mean fluorescence intensity (MFI).
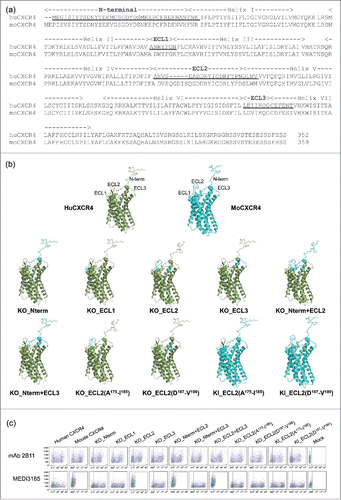
Figure 4. (d) MEDI3185 binding to CXCR4 chimeric variants compared with un-mutated CXCR4. Binding was calculated as % binding when compared with un-mutated human CXCR4 after expression normalization using the following formula: [(MFICXCR4mut MEDI3185/MFICXCR4wt MEDI3185)/(MFICXCR4mut 2B11/MFICXCR4wt 2B11)] *100. Results represent the means of 3 independent experiments.
![Figure 4. (d) MEDI3185 binding to CXCR4 chimeric variants compared with un-mutated CXCR4. Binding was calculated as % binding when compared with un-mutated human CXCR4 after expression normalization using the following formula: [(MFICXCR4mut MEDI3185/MFICXCR4wt MEDI3185)/(MFICXCR4mut 2B11/MFICXCR4wt 2B11)] *100. Results represent the means of 3 independent experiments.](/cms/asset/5f735b31-d666-402f-90c7-fad413245ed8/kmab_a_1113359_f0004b_b.gif)
To further refine the MEDI3185 epitope, Ala scanning mutagenesis was applied to ECL2 (excluding C186). All ECL2 residues were substituted with Ala (except A175 and A180, which were substituted by Asp), alone or in combination, and assessed for MEDI3185 binding (). We also replaced either of the 2 above-mentioned ECL2 portions with unstructured glycine-serine linkers (G3SG3S) in an effort to identify whether well-defined main-chain conformations play a significant role in interacting with MEDI3185. All variants expressed well on the surface of CHO cells as monitored using mAb 2B11 (). Strikingly, none of the Ala or Asp mutations had any significant effect on MEDI3185 binding. MEDI3185 binding was only abolished upon substituting either portion of ECL2 with G3S linkers. These results further confirm that MEDI3185 epitope is ‘dispersed’ throughout the entire ECL2. It also follows that: 1) ECL2 side-chains as a whole do not seem to significantly contribute to MEDI3185 binding; and 2) the main-chains of ECL2 residues likely play a major role in this interaction and their overall conformation is critical to MEDI3185 binding.
Table 1. MEDI3185 binding to CXCR4 ECL2 variants
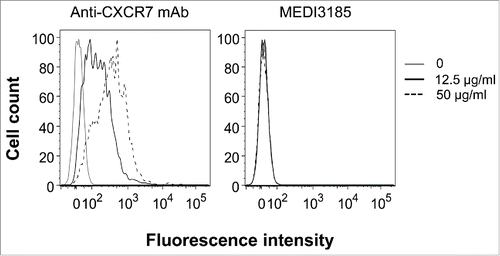
We then examined if MEDI3185 cross-reacts with human CXCR7, a closely related receptor for SDF-1.Citation59 We found that MEDI3185 does not bind to CXCR7-positive (and CXCR4-negative) MCF-7 human breast cancer cells (). Thus, the MEDI3185 epitope appears to be specific to CXCR4.
Figure 5. MEDI3185 does not bind to CXCR7. Human MCF-7 breast cancer cells (CXCR7-positive/CXCR4-negative) were incubated with MEDI3185 or a mouse anti-CXCR7 mAb (clone 11G8).
Homology modeling of MEDI3185
To gain further insight on MEDI3185 interaction with CXCR4, the 3-dimensional structure of the variable domains of MEDI3185 was predicted by homology modeling using antibody structure modeling tools implemented in Discovery Studio. First, framework structures were modeled using the following high homology templates: 1DEE for VL (97% identity), 3EYQ for VH (95% identity) and 1DEE for VL/VH interface (92% identity). Second, CDR conformations were modeled using a set of loop templates, including 2JIX for CDR1L, 2L and 3L (90.9%, 100%, and 77.8% identity, respectively), 8FAB for CDR1H (80% identity), 3EYQ for CDR2H (94.1% identify) and 1ZA3 for CDR3H (30% identity), as summarized in . Third, although structure prediction is fairly accurate for antibody frameworks and CDRs with well-defined canonical structures, modeling CDR3H has proven more challenging and been shown to yield average root mean square deviation values of ˜3.0 Å from the corresponding X-ray crystal structures.Citation60 Therefore, we built this CDR using a template identified using so-called ‘H3 rules’.Citation61,62 These rules are thought to more accurately model CDR3H and rely on the overall analysis of primary sequence, nature and positions of select residues, as well as known structural motifs. We identified 4LKC as the CDR3H template which belongs, along with MEDI3185 CDR3H, to the KG sub-type. Both boast identical length and similar composition (). Using 4LKC as the new structural template, MEDI3185 CDR3H was predicted to form a β-turn structure with a kinked base protruding from a base formed by the other 5 CDRs ().
Figure 6. Modeled 3-dimensional structure of MEDI3185 VH (cyan) and VL (tint) domains. CDRs are shown in orange (VH) and magenta (VL). Intra-chain disulfides are shown as spheres. MEDI3185 exhibits a long CDR3H in a β-hairpin conformation protruding away from the variable domains.
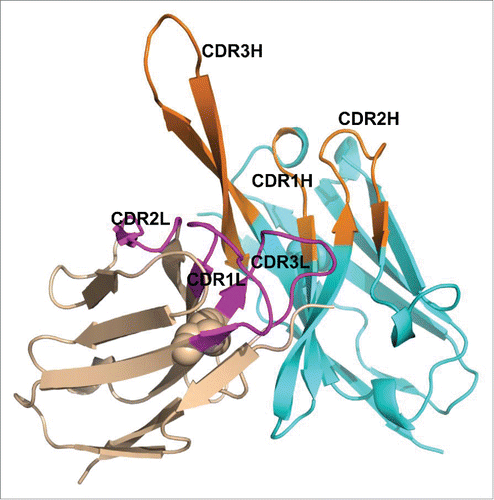
Table 2. CDR templates for MEDI3185 loop modeling
After refinement, the top-ranked model was inspected for clashes between atoms. In such regions, limited minimization was performed for side-chains using CHARMm Citation63,64 implemented in Discovery Studio. The final model was validated using Profiles 3D and Ramachandran plots (96% Ramachandran favored residues).
Mode of interaction between MEDI3185 and CXCR4
Guided by the epitope mapping results, we used the ZDOCKCitation65 and RDOCKCitation66 algorithms to create a model of the MEDI3185/CXCR4 complex. The modeled MEDI3185 Fv structure was docked to human CXCR4 (PDB ID number 3ODU) using ZDOCK. All predicted docked structures were clustered and filtered for poses involving the ECL2 epitope region in the binding interface. The resulting ˜50 poses in the top 2 clusters were manually examined to select poses containing the major MEDI3185 CDR3H paratope in the interface. The qualified 10 poses were further refined and evaluated using RDOCK. All top poses with low RDOCK energies (< -15 kcal.mol-1), including electrostatic and desolvation energies, were advanced for binding interface analysis. The final model was selected to be consistent with all the mutagenesis results.
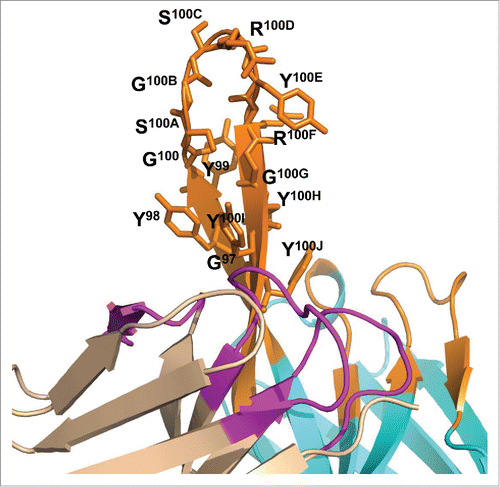
Based on mutagenesis and modeling data, we propose a new mode of interaction for MEDI3185/CXCR4 based on a β-strand/β-strand interaction between CDR3H and ECL2 (). On the epitope side, the entire ECL2 contributes to the interaction with MEDI3185, mostly through its main-chains as described above. This is in very good agreement with our proposed β-strand/β-strand-based mode of binding where main chain/main chain hydrogen bonds constitute the primary contacts. In addition, the conformational complementarity between MEDI3185 CDR3H and CXCR4 ECL2 hairpins may also substantially contribute to the interaction. However, a detailed analysis of the binding interface would require a crystal structure. On the paratope side and as shown in , CDR3H mutations which substantially reduced binding to CXCR4 are localized in the apex of the β-hairpin loop (S100CR100DY100E), the N-terminal strand (G97Y98Y99), and the C-terminal strand (R100FG100GY100H). Therefore, most of CDR3H β-hairpin structure is involved in interacting with CXCR4. These results also agree well with the proposed mode of binding to the extent that although CDR3H side-chain mutations may not break the β-strand/β-strand interaction, they could distort the conformation of the apex loop portion of CDR3H and affect binding to CXCR4.
Figure 7. Three-dimensional representation of the proposed interaction of MEDI3185 with CXCR4. MEDI3185 binds to CXCR4 through a β-strand/β-strand interaction between the 2 corresponding β-hairpin structures on CDR3H (red) and ECL2 (blue). CXCR4 residues D97, D187 and E288 reported to be critical for SDF-1 binding/signalingCitation42 are shown in bright green sticks.
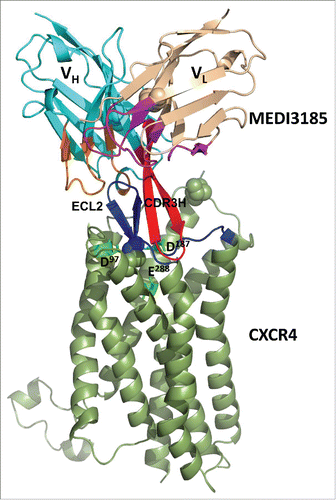
Figure 8. Close-up view of MEDI3185 CDR3H. Mutated residues are shown as sticks.
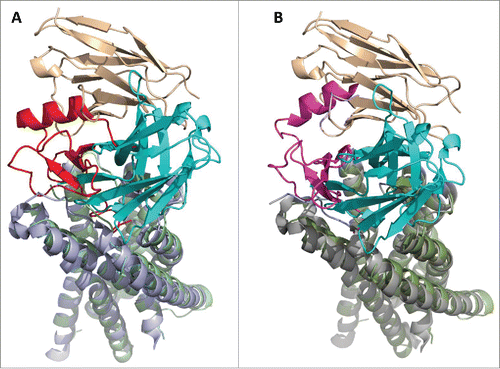
Based on our model, MEDI3185 antagonistic activity is mediated via sterically blocking access of SDF-1 to CXCR4. A “2-site” model has previously been hypothesized for the CXCR4/SDF-1 interaction,Citation40,67-71 whereby CXCR4 N-terminal peptide serves as a docking domain (‘site 1’) for SDF-1, followed by interaction of SDF-1 with the pocket defined by CXCR4 transmembrane helices (‘site 2’). A model of the CXCR4/SDF-1 complex has been proposed using a combination of computational, biochemical and biophysical approaches.Citation72 This model is consistent with the hypothesis of a 1:1 stoichiometry and a 2-site receptor-ligand interaction. Moreover, a recent crystal structure of the complex of CXCR4 bound to a viral chemokine has clearly demonstrated that CXCR4 interacts with the chemokine via its N-terminal peptide (site 1) and transmembrane pocket (site 2),Citation73 further confirming the 2-site receptor-ligand interaction mode. Although MEDI3185 does not bind either of these 2 sites, superimposing the model of the CXCR4/MEDI3185 complex to the structure of CXCR4 bound to the viral chemokine (PDB ID number 4RWS) or SDF-1 (model) shows that MEDI3185 blocks binding of CXCR4 to its ligands via steric hindrance ().
Figure 9. MEDI3185 sterically blocks binding of CXCR4 ligands. The mechanism of action of MEDI3185 is proposed based on superimposing the model of the MEDI3185/CXCR4 (LC tint-HC cyan/olive) complex to (a) the structure of human CXCR4 (light blue) bound to viral chemokine vMIP-II (red; PDB ID number 4RWS)Citation73 or (b) a model of human CXCR4 (gray) bound to SDF-1(magenta).Citation72 Superimpositions were carried out through the common CXCR4 molecules. Upon binding to CXCR4, MEDI3185 interferes with the access of vMIP-II and SDF-1 ligands through both its light and heavy chains.
Discussion
In fine, an actual crystal structure of the MEDI3185/CXCR4 complex will be required to provide a detailed molecular understanding of this interaction. This is a challenging proposition given the GPCR nature of CXCR4. In the meantime, our study represents the first thorough molecular characterization of an interaction between an anti-CXCR4 antagonistic mAb and CXCR4. It also reveals important new findings related to the function and design of therapeutic antibodies against CXCR4.
MEDI3185 contains a long 20-aa CDR3H. Interestingly, one study has described the sequences of a panel of anti-CXCR4 mAbs identified from phage libraries,Citation50 and the most potent antagonist molecules exhibited a long (17 to 18 aa) CDR3H. Additionally, BMS-936564, a human anti-CXCR4 mAb currently in clinical trials also exhibits a long (16 aa) CDR3H (patent entitled “Treatment of hematologic malignancies with an anti-cxcr4 antibody; WO 2013071068A2). Thus, a long CDR3H may be an important attribute for antagonistic anti-CXCR4 antibodies.
Of note, ECL2 appears to be a shared epitope for many reported anti-CXCR4 neutralizing mAbs, and those recognizing other ECLs or the N-terminal peptide exhibit no or only weak activity in inhibiting the CXCR4/SDF-1 signaling pathway and CXCR4-mediated HIV infection.Citation43,48-50,74 However, a unique feature of the MEDI3185 epitope is the lack of a hot-spot on ECL2. It is dispersed throughout the entire ECL2 and involves both the β-hairpin structure (A175-I185) and the loop at its base (D187-V196). Epitopes for previously described mouse antagonistic mAbs 12G5, 701, 708, 716, 717 and 718 all involve 2 hot-spot ECL2 residues at E179 and D181.Citation43,49 These 2 residues are located at the top of the ECL2 β-hairpin structure and are easily accessible by short CDR loops. Indeed, one of these mAbs (12G5) exhibits a short (9 aa) CDR3H.Citation75 In contrast, MEDI3185's long CDR3H allows it to reach the stem region of the ECL2 hairpin and the loop at its base. Furthermore, we also find here that MEDI3185 epitope is resilient to individual and combined Ala mutations. Such a robustness of MEDI3185 epitope may constitute an advantage to efficiently target a wide population of CXCR4 due to its known propensity to exhibit significant conformational heterogeneity.Citation76 Additional binding characterization using CXCR4 from different primary tissues would be required to further validate this hypothesis.
Cell-type dependent glycosylation of CXCR4 also contributes its heterogeneity. CXCR4 contains 2 potential N-glycosylation sites at positions of N11Y12T13 (in the N-terminal peptide) and N176V177S178 (in ECL2). Mutations of the N11 glycation site was shown to decrease the binding of the N-terminal peptide-recognizing mAb 2B11.Citation77 However, binding of ECL2-recognizing mAb 12G5 was not affected by mutations of either N11 or N176.Citation77,78 Similarly to 12G5, we expect little to no impact of these different N-glycosylation profiles on MEDI3185 binding because MEDI3185 only binds to ECL2 and mutating N176VSE179 to a stretch of Ala did not affect the binding of MEDI3185 (). Here again, additional binding experiments using cells isolated from primary tissues would be needed to confirm.
Some GPCR drugs act as inverse agonists rather than neutral antagonistsCitation79 and exhibit potential therapeutic benefits when compared with neutral antagonists in diseases where increased levels of GPCR basal activity is seen. Although it is unlikely that MEDI3185 functions as an inverse agonist in light of its binding site and affinity, we cannot rule out this possibility based on the data presented here. Full functional characterization of MEDI3185 in terms of inhibiting CXCR4/SDF-1 dependent tumor cell signaling, migration, and proliferation will be described elsewhere (manuscript in preparation).
Materials and Methods
Construction and expression of CXCR4 and MEDI3185 variants
DNA encoding full length human and mouse CXCR4 (NCBI reference NP_003458.1 and NP_034041.2, respectively) as well as MEDI3185 (MedImmune, human IgG1/κ) were generated at MedImmune. CXCR4 and MEDI3185 variants were generated and assembled using PCR by overlap extension. Human and mouse CXCR4 and their variants were cloned into the mammalian expression vector pcDNA3.1 (Invitrogen). MEDI3185 wild-type and variants were cloned into an Orip/EBNA-1-based episomal mammalian expression plasmid, pOE.Citation80 CHO cells were transiently transfected with CXCR4 constructs using Lipofectamine® LTX reagent with PLUS™ reagent (Invitrogen) according to the manufacturer's instructions, and harvested for flow cytometry characterization 24 h post-transfection. Constructs encoding MEDI3185 were also transfected into CHO cells using Lipofectamine® LTX reagent with PLUS™ reagent, and conditioned media was harvested 5 days post-transfection.
Binding of MEDI3185 to CXCR4 variants
About 106 CHO cells were transfected with the various CXCR4 constructs, then incubated with either 0.3 μg/ml or a serial dilutions (0.6-0.009 µg/ml) of MEDI3185 in 50 µl phosphate-buffered saline (PBS) containing 1% BSA for 30 min on ice. Cells were washed 3 times with 200 µl ice-cold PBS, and incubated with 1 μg/ml of an anti-human IgG antibody conjugated to FITC (Invitrogen) in 50 µl PBS containing 1% BSA for 30 min on ice. Expression of CXCR4 variants was monitored by incubating the cells with 10 µg/ml APC-conjugated mAb 2B11 (eBioscience) in 50 µl PBS containing 1% BSA for 30 min on ice, and washing 3 times with 200 µl ice-cold PBS. All samples were analyzed using a LSRII flow cytometer (BD Biosciences). KD values were calculated using a non-linear regression with the GraphPad Prism software.
Competition binding between MEDI3185 and AMD3100
Competition between MEDI3185 and AMD3100 (R&D System) for binding to CXCR4 was assessed by incubating CXCR4-positive Jurkat cells (ATCC) with 1 nM MEDI3185 in the presence of 10 μM AMD3100. This was followed by incubation with 1 μg/ml of an anti-human IgG antibody conjugated to FITC (Invitrogen) in 50 µl PBS containing 1% BSA for 30 min on ice and 3 washes with 200 µl ice-cold PBS. MEDI3185 binding was analyzed using a LSRII flow cytometer (BD Biosciences).
Binding of MEDI3185 variants to CXCR4
Conditioned media from MEDI3185-expressing CHO cells was submitted to IgG quantification using a ForteBIO Octet QK 384. Supernatants were diluted 4-fold with growth media (Invitrogen) and transferred into a 384-well tilted bottom plate (ForteBIO). Purified MEDI3185 was diluted to 100 μg/ml in growth media and a standard curve was generated using serial 2-fold dilutions. Protein A biosensors (ForteBIO) were pre-conditioned with growth media for 10 min and diluted supernatants loaded onto the sensors for ˜300 sec. Data analysis was carried out using the QK384 Analysis software (ForteBIO). After concentration normalization, MEDI3185 variants were incubated with 293X cells (expressing endogenous CXCR4) at 0.1 and 0.03 µg/ml in 50 µl PBS containing 1% BSA for 30 min on ice. Cells were washed 3 times with 200 µl ice-cold PBS and incubated with 1 μg/ml of an anti-human IgG antibody conjugated to FITC (Invitrogen) in 50 µl PBS containing 1% BSA for 30 min on ice. Cells were then washed 3 times with 200 µl ice-cold PBS. Bound antibodies were detected using a LSRII flow cytometer.
Binding of MEDI3185 to CXCR7
About 106 human MCF-7 breast cancer cells (ATCC) were incubated on ice with MEDI3185 at 12.5 or 50 μg/ml in 50 µl PBS containing 1% BSA for 30 min in a 96-well plate. Cells were washed 3 times with 200 µl ice-cold PBS and incubated with 1 μg/ml of an anti-human IgG antibody conjugated to FITC (Invitrogen) in 50 µl PBS containing 1% BSA for 30 min on ice. CXCR7 expression was checked by incubating MCF-7 cells with 12.5 or 50 μg/ml of anti-CXCR7 mAb clone 11G8 (R&D Systems) in 50 µl PBS containing 1% BSA for 30 min on ice, followed by 3 washes with 200 µl ice-cold PBS then incubation with 1 μg/ml of an anti-mouse-IgG mAb conjugated to phycoerythrin (PE; Invitrogen) on ice for 30 min. Cells were then washed 3 times with 200 µl ice-cold PBS. All samples were analyzed using a LSRII flow cytometer.
MEDI3185 structure modeling
The structure of MEDI3185 variable domains was predicted using Discovery Studio 3.5 (DS 3.5; Biovia). Default antibody structure modeling protocols were used to model the frameworks as well as CDRL1, L2, L3, H1 and H2. Briefly, a BLAST search was performed against the protein data bankCitation81 to identify framework templates (one each for VH, VL and VH/VL interface) exhibiting the highest sequence homology to MEDI3185. 100 models were then constructed through homology modeling using these 3 structural templates. The top scored framework model with the lowest probability density function (PDF) energy was further selected for modeling the CDR loops. CDRs were built by homology modeling using CDR templates sharing the highest sequence identity when compared with MEDI3185 CDRs. In particular, CDR3H was rebuilt by incorporating so-called ‘H3 rules’ (see below). The top ranked model was then inspected for clashes between atoms, in which case limited minimization was performed for side-chains using CHARMm. The quality of the MEDI3185 model was validated using Profiles 3D and Ramachandran plots of DS 3.5. Illustrations were prepared using PyMOL (Schrödinger).
Docking
ZDOCK in DS 3.5 was used to dock human CXCR4 to the MEDI3185 model. CXCR4 coordinates were prepared for docking using PDB ID number 3ODU40 and the protein preparation tool in DS3.5. CHARMm force fieldCitation63,64 was applied throughout the simulation. Rigid-body docking was performed at a 6° angular step size and clustered for the top 2000 poses. All poses from ZDOCK were processed by filtering for those containing CXCR4 ECL2 within 5 Å to MEDI3185. Clusters with the highest density of poses were further considered and went through manual examination to deselect those involving mainly MEDI3185 framework regions in binding. Selected poses were then refined and evaluated using RDOCK and the top ones exhibiting low RDOCK energies were advanced for binding interface analysis. All docking calculations were made with DS 3.5.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Vaheh Oganesyan, Keven Huang, and Adeela Kamal for insightful discussions and providing reagents.
References
- Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996; 272:872–77; PMID:8629022; http://dx.doi.org/10.1126/science.272.5263.872
- Bleul CC, Farzan M, Choe H, Parolin C, ClarkLewis I, Sodroski J, Springer TA. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 1996; 382:829–33; PMID:8752280; http://dx.doi.org/10.1038/382829a0
- Loetscher M, Geiser T, O'Reilly T, Zwahlen R, Baggiolini M, Moser B. Cloning of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in leukocytes. J Biol Chem 1994; 269:232–37; PMID:8276799
- Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998; 393:595–99; PMID:9634238; http://dx.doi.org/10.1038/31269
- Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, Bronson RT, Springer TA. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. PNAS USA 1998; 95:9448–53; PMID:9689100; http://dx.doi.org/10.1073/pnas.95.16.9448
- McGrath KE, Koniski AD, Maltby KM, McGann JK, Palis J. Embryonic expression and function of the chemokine SDF-1 and its receptor, CXCR4. Dev Biol 1999; 213:442–56; PMID:10479460; http://dx.doi.org/10.1006/dbio.1999.9405
- Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, Yoshida N, Kikutani H, Kishimoto T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996; 382:635–38; PMID:8757135; http://dx.doi.org/10.1038/382635a0
- Murphy PM. The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol 1994; 12:593–633; PMID:8011292; http://dx.doi.org/10.1146/annurev.iy.12.040194.003113
- Doranz BJ, Berson JF, Rucker J. Doms RW. Chemokine receptors as fusion cofactors for human immunodeficiency virus type 1 (HIV-1). Immunol Res 1997; 16:15–28; PMID:9048206; http://dx.doi.org/10.1007/BF02786321
- Nanki T, Hayashida K, El-Gabalawy HS, Suson S, Shi K, Girschick HJ, Yavuz S, Lipsky PE. Stromal cell-derived factor-1-CXC chemokine receptor 4 interactions play a central role in CD4+ T cell accumulation in rheumatoid arthritis synovium. J Immunol 2000; 165:6590–98; PMID:11086103; http://dx.doi.org/10.4049/jimmunol.165.11.6590
- Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001; 410:50–56; PMID:11242036; http://dx.doi.org/10.1038/35065016
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer 2004; 4:540–50; PMID:15229479; http://dx.doi.org/10.1038/nrc1388
- Zlotnik A. New insights on the role of CXCR4 in cancer metastasis. J Pathol 2008; 215:211–13; PMID:18523970; http://dx.doi.org/10.1002/path.2350
- Fulton AM. The chemokine receptors CXCR4 and CXCR3 in cancer. Curr Oncol Rep 2009; 11:125–31; PMID:19216844; http://dx.doi.org/10.1007/s11912-009-0019-1
- Teicher BA. Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res 2010; 16:2927–31; PMID:20484021; http://dx.doi.org/10.1158/1078-0432.CCR-09-2329
- Balkwill F. The significance of cancer cell expression of the chemokine receptor CXCR4. Semin Cancer Biol 2004; 14:171–79; PMID:15246052; http://dx.doi.org/10.1016/j.semcancer.2003.10.003
- Tamamura H, Fujii N. The therapeutic potential of CXCR4 antagonists in the treatment of HIV infection, cancer metastasis and rheumatoid arthritis. Expert Opin. Ther. Targets 2005; 9:1267–82
- Patrussi L, Baldari CT. The CXCL12/CXCR4 axis as a therapeutic target in cancer and HIV-1 infection. Curr Med Chem 2011; 18:497–512; PMID:21143114; http://dx.doi.org/10.2174/092986711794480159
- Weitzenfeld P, Ben-Baruch A. The chemokine system, and its CCR5 and CXCR4 receptors, as potential targets for personalized therapy in cancer. Cancer Lett 2014; 352:36–53; PMID:24141062; http://dx.doi.org/10.1016/j.canlet.2013.10.006
- Kuhne MR, Mulvey T, Belanger B, Chen S, Pan C, Chong C, Cao F, Niekro W, Kempe T, Henning KA, et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res 2013; 19:357–66; PMID:23213054; http://dx.doi.org/10.1158/1078-0432.CCR-12-2333
- Kamal A, Steiner P, Wang Y, Wetzel L, Mazzola A, Passino M, McDermott B, Huang K, Peng L, Rebelatto M, et al. (2013) AACR Meeting Abstracts abstract 5462.
- Schols D, Struyf S, Van Damme J, Este JA, Henson G, De Clercq E. Inhibition of T-tropic HIV strains by selective antagonization of the chemokine receptor CXCR4. J Exp Med 1997; 186:1383–88; PMID:9334378; http://dx.doi.org/10.1084/jem.186.8.1383
- De Clercq E. The bicyclam AMD3100 story. Nat Rev Drug Discov 2003; 2:581–87; PMID:12815382; http://dx.doi.org/10.1038/nrd1134
- Donzella GA, Schols D, Lin SW, Este JA, Nagashima KA, Maddon PJ, Allaway GP, Sakmar TP, Henson G, De Clercq E, et al. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med 1998; 4:72–7; PMID:9427609; http://dx.doi.org/10.1038/nm0198-072
- Matthys P, Hatse S, Vermeire K, Wuyts A, Bridger G, Henson GW, De Clercq E, Billiau A, Schols D. AMD3100, a potent and specific antagonist of the stromal cell-derived factor-1 chemokine receptor CXCR4, inhibits autoimmune joint inflammation in IFN-gamma receptor-deficient mice. J Immunol 2001; 167:4686–92; PMID:11591799; http://dx.doi.org/10.4049/jimmunol.167.8.4686
- Lukacs NW, Berlin A, Schols D, Skerlj RT, Bridger GJ. AMD3100, a CxCR4 antagonist, attenuates allergic lung inflammation and airway hyperreactivity. Am J Pathol 2002; 160:1353–60; PMID:11943720; http://dx.doi.org/10.1016/S0002-9440(10)62562-X
- Liles WC, Broxmeyer HE, Rodger E, Wood B, Hubel K, Cooper S, Hangoc G, Bridger GJ, Henson GW, Calandra G, et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood 2003; 102:2728–30; PMID:12855591; http://dx.doi.org/10.1182/blood-2003-02-0663
- Domanska UM, Timmer-Bosscha H, Nagengast WB, Oude Munnink TH, Kruizinga RC, Ananias HJ, Kliphuis NM, Huls G, De Vries EG, de Jong IJ, et al. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia 2012; 14:709–18; PMID:22952424; http://dx.doi.org/10.1593/neo.12324
- De Clercq E. Recent advances on the use of the CXCR4 antagonist plerixafor (AMD3100, Mozobil) and potential of other CXCR4 antagonists as stem cell mobilizers. Pharmacol Ther 2010; 128:509–18; PMID:20826182; http://dx.doi.org/10.1016/j.pharmthera.2010.08.009
- Kessans MR, Gatesman ML, Kockler DR. Plerixafor: a peripheral blood stem cell mobilizer. Pharmacotherapy 2010; 30:485–92; PMID:20411999; http://dx.doi.org/10.1592/phco.30.5.485
- Choi HY, Yong CS, Yoo BK. Plerixafor for stem cell mobilization in patients with non-Hodgkin's lymphoma and multiple myeloma. Ann Pharmacother 2010; 44:117–26; PMID:20009003; http://dx.doi.org/10.1345/aph.1M380
- Murakami T, Nakajima T, Koyanagi Y, Tachibana K, Fujii N, Tamamura H, Yoshida N, Waki M, Matsumoto A, Yoshie O, et al. A small molecule CXCR4 inhibitor that blocks T cell line-tropic HIV-1 infection. J Exp Med 1997; 186:1389–93; PMID:9334379; http://dx.doi.org/10.1084/jem.186.8.1389
- Arakaki R, Tamamura H, Premanathan M, Kanbara K, Ramanan S, Mochizuki K, Baba M, Fujii N, Nakashima H. T134, a small-molecule CXCR4 inhibitor, has no cross-drug resistance with AMD3100, a CXCR4 antagonist with a different structure. J Virol 1999; 73:1719–23; PMID:9882387
- Tamamura H, Hori A, Kanzaki N, Hiramatsu K, Mizumoto M, Nakashima H, Yamamoto N, Otaka A, Fujii N. T140 analogs as CXCR4 antagonists identified as anti-metastatic agents in the treatment of breast cancer. FEBS Lett 2003; 550:79–83; PMID:12935890; http://dx.doi.org/10.1016/S0014-5793(03)00824-X
- Tamamura H, Hiramatsu K, Mizumoto M, Ueda S, Kusano S, Terakubo S, Akamatsu M, Yamamoto N, Trent JO, Wang ZP, et al. Enhancement of the T140-based pharmacophores leads to the development of more potent and bio-stable CXCR4 antagonists. Org Biomol Chem 2003; 1:3663–69; PMID:14649897; http://dx.doi.org/10.1039/b306613b
- Tamamura H, Esaka A, Ogawa T, Araki T, Ueda S, Wang Z, Trent JO, Tsutsumi H, Masuno H, Nakashima H, et al. Structure-activity relationship studies on CXCR4 antagonists having cyclic pentapeptide scaffolds. Org Biomol Chem 2005; 3:4392–94; PMID:16327900; http://dx.doi.org/10.1039/b513145f
- Rosenkilde MM, Gerlach LO, Jakobsen JS, Skerlj RT, Bridger GJ, Schwartz TW. Molecular mechanism of AMD3100 antagonism in the CXCR4 receptor: transfer of binding site to the CXCR3 receptor. J Biol Chem 2004; 279:3033–41; PMID:14585837; http://dx.doi.org/10.1074/jbc.M309546200
- Tamamura H, Xu Y, Hattori T, Zhang X, Arakaki R, Kanbara K, Omagari A, Otaka A, Ibuka T, Yamamoto N, et al. A low-molecular-weight inhibitor against the chemokine receptor CXCR4: a strong anti-HIV peptide T140. Biochem Biophys Res Commun 1998; 253:877–82; PMID:9918823; http://dx.doi.org/10.1006/bbrc.1998.9871
- Kawatkar SP, Yan M, Gevariya H, Lim MY, Eisold S, Zhu X, Huang Z, An J. Computational analysis of the structural mechanism of inhibition of chemokine receptor CXCR4 by small molecule antagonists. Exp Biol Med (Maywood) 2011; 236:844–50; PMID:21697335; http://dx.doi.org/10.1258/ebm.2011.010345
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010; 330:1066–71; PMID:20929726; http://dx.doi.org/10.1126/science.1194396
- Yoshikawa Y, Kobayashi K, Oishi S, Fujii N, Furuya T. Molecular modeling study of cyclic pentapeptide CXCR4 antagonists: new insight into CXCR4-FC131 interactions. Bioorg Med Chem Lett 2012; 22:2146–50; PMID:22365757; http://dx.doi.org/10.1016/j.bmcl.2012.01.134
- Brelot A, Heveker N, Montes M, Alizon M. Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J Biol Chem 2000; 275:23736–44; PMID:10825158; http://dx.doi.org/10.1074/jbc.M000776200
- Brelot A, Heveker N, Adema K, Hosie MJ, Willett B, Alizon M. Effect of mutations in the second extracellular loop of CXCR4 on its utilization by human and feline immunodeficiency viruses. J Virol 1999; 73:2576–86; PMID:10074102
- Bertolini F, Dell'Agnola C, Mancuso P, Rabascio C, Burlini A, Monestiroli S, Gobbi A, Pruneri G, Martinelli G. CXCR4 neutralization, a novel therapeutic approach for non-Hodgkin's lymphoma. Cancer Res 2002; 62:3106–12; PMID:12036921
- Engl T, Relja B, Marian D, Blumenberg C, Muller I, Beecken WD, Jones J, Ringel EM, Bereiter-Hahn J, Jonas D, et al. CXCR4 chemokine receptor mediates prostate tumor cell adhesion through alpha5 and beta3 integrins. Neoplasia 2006; 8:290–301; PMID:16756721; http://dx.doi.org/10.1593/neo.05694
- Gelmini S, Mangoni M, Castiglione F, Beltrami C, Pieralli A, Andersson KL, Fambrini M, Taddei GL, Serio M, Orlando C. The CXCR4/CXCL12 axis in endometrial cancer. Clin Exp Metastasis 2009; 26:261–68; http://dx.doi.org/10.1007/s10585-009-9240-4
- Cheng Z, Zhou S, Wang X, Xie F, Wu H, Liu G, Wang Q, Chen Y, Hu Y, Lu B, et al. Characterization and application of two novel monoclonal antibodies against human CXCR4: cell proliferation and migration regulation for glioma cell line in vitro by CXCR4/SDF-1alpha signal. Hybridoma (Larchmt) 2009; 8:33–41; http://dx.doi.org/10.1089/hyb.2008.0069
- Tanaka R, Yoshida A, Murakami T, Baba E, Lichtenfeld J, Omori T, Kimura T, Tsurutani N, Fujii N, Wang ZX, et al. Unique monoclonal antibody recognizing the third extracellular loop of CXCR4 induces lymphocyte agglutination and enhances human immunodeficiency virus type 1-mediated syncytium formation and productive infection. J Virol 2001; 75:11534–43; PMID:11689635; http://dx.doi.org/10.1128/JVI.75.23.11534-11543.2001
- Carnec X, Quan L, Olson WC, Hazan U, Dragic T. Anti-CXCR4 monoclonal antibodies recognizing overlapping epitopes differ significantly in their ability to inhibit entry of human immunodeficiency virus type 1. J Virol 2005; 79:1930–33; PMID:15650218; http://dx.doi.org/10.1128/JVI.79.3.1930-1933.2005
- Xu C, Sui J, Tao H, Zhu Q, Marasco WA. Human anti-CXCR4 antibodies undergo VH replacement, exhibit functional V-region sulfation, and define CXCR4 antigenic heterogeneity. J Immunol 2007; 179:2408–18; PMID:17675502; http://dx.doi.org/10.4049/jimmunol.179.4.2408
- Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of immunological interest. U.S. Public Health Service, National Institutes of Health, Washington, DC 1997.
- Zemlin M, Klinger M, Link J, Zemlin C, Bauer K, Engler JA, Schroeder HW, Kirkham PM. Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. J Mol Biol 2003; 334:733–49; PMID:14636599; http://dx.doi.org/10.1016/j.jmb.2003.10.007
- Chothia C, Lesk AM, Tramontano A, Levitt M, Smith-Gill SJ, Air G, Sheriff S, Padlan EA, Davies D, Tulip WR, et al. Conformations of immunoglobulin hypervariable regions. Nature 1989; 342:877–83; PMID:2687698; http://dx.doi.org/10.1038/342877a0
- Chothia C, Lesk AM, Gherardi E, Tomlinson IM, Walter G, Marks JD, Llewelyn MB, Winter G. Structural repertoire of the human VH segments. J Mol Biol 1992; 227:799–817; PMID:1404389; http://dx.doi.org/10.1016/0022-2836(92)90224-8
- Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol 1997; 273:927–48; PMID:9367782; http://dx.doi.org/10.1006/jmbi.1997.1354
- Foote J, Winter G. Antibody Framework Residues Affecting the Conformation of the Hypervariable Loops. J Mol Biol 1992; 224:487–99; PMID:1560463; http://dx.doi.org/10.1016/0022-2836(92)91010-M
- Forster R, Kremmer E, Schubel A, Breitfeld D, Kleinschmidt A, Nerl C, Bernhardt G, Lipp M. Intracellular and surface expression of the HIV-1 coreceptor CXCR4/fusin on various leukocyte subsets: rapid internalization and recycling upon activation. J Immunol 1998; 160:1522–31; PMID:9570576
- Gerlach LO, Skerlj RT, Bridger GJ, Schwartz TW. Molecular interactions of cyclam and bicyclam non-peptide antagonists with the CXCR4 chemokine receptor. J Biol Chem 2001; 276:14153–60; PMID:11154697
- Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold M, Sunshine MJ, Littman DR, Kuo CJ, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med 2006; 203:2201–13; PMID:16940167; http://dx.doi.org/10.1084/jem.20052144
- Almagro JC, Beavers MP, Hernandez-Guzman F, Maier J, Shaulsky J, Butenhof K, Labute P, Thorsteinson N, Kelly K, Teplyakov A, et al. Antibody modeling assessment. Proteins 2011; 79:3050–66; PMID:21935986; http://dx.doi.org/10.1002/prot.23130
- Shirai H, Kidera A, Nakamura H. H3-rules: identification of CDR-H3 structures in antibodies. FEBS Lett 1999; 455:188–97; PMID:10428499; http://dx.doi.org/10.1016/S0014-5793(99)00821-2
- Kuroda D, Shirai H, Kobori M, Nakamura H. Structural classification of CDR-H3 revisited: a lesson in antibody modeling. Proteins 2008; 73:608–20; PMID:18473362; http://dx.doi.org/10.1002/prot.22087
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. Charmm - a Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comp Chem 1983; 4:187–217; http://dx.doi.org/10.1002/jcc.540040211
- Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: The Biomolecular Simulation Program. J Comp Chem 2009; 30:1545–1614; http://dx.doi.org/10.1002/jcc.21287
- Chen R, Li L, Weng Z. ZDOCK: An Initial-stage Protein-Docking algorithm. Proteins 2003; 52: 80–7; PMID:12784371; http://dx.doi.org/10.1002/prot.10389
- Li L, Chen R, Weng Z. RDOCK: refinement of rigid-body protein docking predictions. Proteins 2003; 53:693–707; PMID:14579360; http://dx.doi.org/10.1002/prot.10460
- Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, Virelizier JL, Baggiolini M, Sykes BD, Clark-Lewis I. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J 1997; 16:6996–7007; PMID:9384579; http://dx.doi.org/10.1093/emboj/16.23.6996
- Dealwis C, Fernandez EJ, Thompson DA, Simon RJ, Siani MA, Lolis E. Crystal structure of chemically synthesized ; N33A stromal cell-derived factor 1alpha, a potent ligand for the HIV-1 "fusin" coreceptor. PNAS USA 1998; 95:6941–46; PMID:9618518; http://dx.doi.org/10.1073/pnas.95.12.6941
- Gupta SK, Pillarisetti K, Thomas RA, Aiyar N. Pharmacological evidence for complex and multiple site interaction of CXCR4 with SDF-1alpha: implications for development of selective CXCR4 antagonists. Immunol Lett 2001; 78:29–34; PMID:11470148; http://dx.doi.org/10.1016/S0165-2478(01)00228-0
- Clark-Lewis I, Kim KS, Rajarathnam K, Gong JH, Dewald B, Moser B, Baggiolini M, Sykes BD. Structure-activity relationships of chemokines. J Leukoc Biol 1995; 57:703–11; PMID:7759949
- Wells TN, Power CA, Lusti-Narasimhan M, Hoogewerf AJ, Cooke RM, Chung CW, Peitsch MC, Proudfoot AE. Selectivity and antagonism of chemokine receptors. J Leukoc Biol 1996; 59:53–60; PMID:8558067
- Kufareva I, Stephens BS, Holden LG, Qin L, Zhao C, Kawamura T, Abagyan R, Handel TM. Stoichiometry and geometry of the CXC chemokine receptor 4 complex with CXC ligand 12: Molecular modeling and experimental validation. PNAS 2014; 111:E5363–E5372; PMID:25468967; http://dx.doi.org/10.1073/pnas.1417037111
- Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, Fenalti G, Wu H, Han GW, Cherezov V, et al. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 2015; 347:1117–22; PMID:25612609; http://dx.doi.org/10.1126/science.1261064
- Jähnichen S, Blanchetot C, Maussang D, Gonzalez-Pajuelo M, Chow KY, Bosch L, De Vrieze S, Serruys B, Ulrichts H, Vandevelde W, et al. CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. PNAS 2010; 107:20565–70; http://dx.doi.org/10.1073/pnas.1012865107
- BouHamdan M, Strayer DS, Wei D, Mukhtar M, Duan LX, Hoxie J, Pomerantz RJ. Inhibition of HIV-1 infection by down-regulation of the CXCR4 co-receptor using an intracellular single chain variable fragment against CXCR4. Gene Ther 2001; 8:408–18; PMID:11313818; http://dx.doi.org/10.1038/sj.gt.3301411
- Baribaud F, Edwards TG, Sharron M, Brelot A, Heveker N, Price K, Mortari F, Alizon M, Tsang M, Doms RW. Antigenically distinct conformations of CXCR4. J Virol 2001; 75:8957–67; PMID:11533159; http://dx.doi.org/10.1128/JVI.75.19.8957-8967.2001
- Huskens D, Princen K, Schreiber M, Schols D. The role of N-glycosylation sites on the CXCR4 receptor for CXCL-12 binding and signaling and X4 HIV-1 viral infectivity. Virology 2007; 363:280–7; PMID:17331556; http://dx.doi.org/10.1016/j.virol.2007.01.031
- Chabot DJ, Chen H, Dimitrov DS, Broder CC. N-Linked Glycosylation of CXCR4 Masks Coreceptor Function for CCR5-Dependent Human Immunodeficiency Virus Type 1 Isolates. J Virol 2000; 74:4404–13; PMID:10756055; http://dx.doi.org/10.1128/JVI.74.9.4404-4413.2000
- Milligan G. Constitutive activity and inverse agonists of G protein-coupled receptors: a current perspective. Mol Pharmacol 2003; 64:1271–6; PMID:14645655; http://dx.doi.org/10.1124/mol.64.6.1271
- Dimasi N, Gao C, Fleming R, Woods RM, Yao XT, Shirinian L, Kiener PA, Wu H. The design and characterization of oligospecific antibodies for simultaneous targeting of multiple disease mediators. J Mol Biol 2009; 393:672–92; PMID:19699208; http://dx.doi.org/10.1016/j.jmb.2009.08.032
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res 2000; 28:235–42; http://dx.doi.org/10.1093/nar/28.1.235