
ABSTRACT
CD73 (ecto-5′-nucleotidase) has recently been established as a promising immuno-oncology target. Given its role in activating purinergic signaling pathways to elicit immune suppression, antagonizing CD73 (i.e., releasing the brake) offers a complimentary pathway to inducing anti-tumor immune responses. Here, we describe the mechanistic activity of a new clinical therapeutic, MEDI9447, a human monoclonal antibody that non-competitively inhibits CD73 activity. Epitope mapping, structural, and mechanistic studies revealed that MEDI9447 antagonizes CD73 through dual mechanisms of inter-CD73 dimer crosslinking and/or steric blocking that prevent CD73 from adopting a catalytically active conformation. To our knowledge, this is the first report of an antibody that inhibits an enzyme's function through 2 distinct modes of action. These results provide a finely mapped epitope that can be targeted for selective, potent, and non-competitive inhibition of CD73, as well as establish a strategy for inhibiting enzymes that function in both membrane-bound and soluble states.
Abbreviatons
| mAb | = | monoclonal antibody |
| kDa | = | kilodalton |
| APCP | = | adenosine 5′-(α,β-methylene) diphosphate |
| sCD73 | = | soluble CD73 |
| HDX | = | hydrogen deuterium exchange |
| aa | = | amino acid |
| CDR | = | complementarity-determining region |
| Fab | = | fragment antigen binding |
| Fv | = | variable fragment |
| VH | = | heavy chain variable domain |
| VL | = | light chain variable domain |
| xFd | = | antibody Fd antibody |
| pFab | = | polyclonal Fab |
| BLI | = | biolayer interferometry |
| SEC-MALS | = | size-exclusion chromatography multi-angle light scattering |
Introduction
CD73 is a highly conserved ecto-nucleotidase that catalyzes the dephosphorylation of a subset of 5′ nucleotides.Citation1 Its primary substrate is 5′AMP, making it the predominant enzyme to produce extracellular adenosine.Citation1,2 In recent years, studies have revealed CD73 plays a significant role in promoting cancer progression by upregulating adenosine signaling, a well-established mechanism of inhibiting immunosurveillance against tumor cells.Citation3-5 Adenosine signaling through A2a receptors on CD4+ and CD8+ T lymphocytes and natural killer cells leads to the suppression of effector functions.Citation6,7 Furthermore, it has been shown that binding of adenosine to A2a and A2b receptors can downregulate macrophage phagocytosis, inhibit proinflammatory cytokine release, upregulate pro-angiogenic factors, and yield aberrantly differentiated dendritic cells producing pro-tumorigenic molecules.Citation8-11 Moreover, many cancer cells overexpress CD73, which exacerbates immune suppression in the tumor microenvironment.Citation12,13 These studies, among others, highlight the therapeutic potential of blocking CD73 hydrolysis of AMP as a means to de-repress anti-tumor immune activity. Indeed, inhibiting CD73 function in vivo has been shown to inhibit tumor growth, metastasis, and neoangiogenesis.Citation14–18
CD73 exists as a homodimer bound to the outer leaflet of the plasma membrane via a glycophosphatidylinositol (GPI) anchor, although it can be shed to yield a soluble form that is also hydrolytically active.Citation19,20 Each ˜65 kgdalton (kDa) monomer is composed of glycosylated N- and C-terminal domains joined by a highly flexible α helical linker.Citation21 In vitro structure-function studies demonstrated that the hydrolysis of AMP by CD73 requires that the enzyme cycle through open and closed conformational states.Citation21-23 This structural transition involves an extensive rotation of the N-terminal domain, described as a “ball-and-socket” motion, in order to form the catalytically active, closed conformer.Citation21,24 Published structural data have facilitated the selection and design of small molecule chemical inhibitors of CD73, which include both naturally occurring and synthetic compounds such as nucleotide di- or triphosphates, alkaloids, and sulfa drugs.Citation2,25-28 Many of these, including the non-hydrolyzable ADP analog adenosine 5′-(α,β-methylene) diphosphate (APCP), are competitive inhibitors that resemble the structure of AMP.Citation26 Others act indirectly to modulate CD73 function or have unknown mechanisms.Citation29 Although it should be possible to develop small molecules targeting CD73 as therapeutics, to date none have progressed to clinical studies, possibly because of cytotoxicity, off-target effects, lack of selectivity or other undesirable pharmacological properties that will need to be addressed in future drug design.Citation30
Monoclonal antibodies (mAbs) represent an attractive alternative to small molecules for inhibiting CD73. Several studies have demonstrated the efficacy of mAbs in antagonizing CD73 function as a means to inhibit tumor growth.Citation14,31-34 While the specific effects of these antibodies on CD73 vary, presumably due to differences in the mAb interaction sites with CD73, they can induce enzyme internalization, shedding, or inhibit AMP hydrolysis.Citation20,31-33 We previously described anti-CD73 antibodies isolated from phenotypic screens that both induce CD73 internalization and enzyme inhibition, and demonstrated one of these mAbs inhibits xenograft tumor growth, likely through these mechanisms.Citation32,35
Currently, there is a need for selective and potent inhibitors of CD73 that can be developed for clinical application. In this study, we describe MEDI9447, a human, high-affinity antagonistic antibody that inhibits CD73 hydrolysis of AMP. In vivo, MEDI9447 reduces adenosine-mediated suppression of immune effector cells and inhibits tumor growth (Hay et al unpublished data).Citation36 Using hydrogen-deuterium exchange mass spectrometry and mutagenesis strategies, we defined the epitope of MEDI9447 and examined the potential effects of antibody binding on the global CD73 structure. The antibody binds to a site in the N-terminal domain of CD73, which results in non-competitive inhibition of AMP hydrolysis. Remarkably, the epitope is positioned such that antibody binding impedes the conversion of both membrane-associated and soluble CD73 from the open conformer to the catalytically active, closed state. This unique mode of action is conferred through 2 different mechanisms mediated by the valency of the antibody interaction with CD73 in its soluble or membrane-bound state.
Results
Inhibition of CD73 enzyme activity by MEDI9447
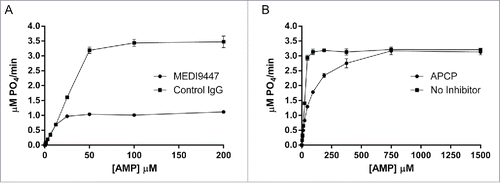
MEDI9447 was discovered and optimized for CD73 binding and neutralization using established phage display methodology. To characterize the inhibitory activity of MEDI9447 IgG, we examined the kinetics of soluble CD73 (sCD73) AMP hydrolysis in the presence of either MEDI9447 or the non-hydrolyzable CD73 inhibitor, APCP using the Malachite Green assay. MEDI9447 non-competitively inhibited sCD73, as was evidenced by decreased Vmax (4.6 ± 0 . vs 1.2 ± 0 .0 μM P04/min) and constant Km (). In contrast, the competitive inhibitor of CD73 APCP did not decrease Vmax (3.4 ± 0 .0 vs 3.50±0 .1 μM P04/min) but increased the Km (75.9 ± 3 .4 vs 26.0±3 .9 μM), data in agreement with previous findings ().Citation37 These results show that MEDI9447 does not prevent the binding of the AMP substrate, yet blocks the ability of sCD73 to hydrolyze AMP.
Figure 1. MEDI9447 is a non-competitive inhibitor of CD73 hydrolysis of AMP. (A) The enzyme kinetics of CD73 phosphohydrolysis of AMP were measured in the presence of MEDI9447 or an isotype-matched control mAb using the Malachite Green assay. Increasing AMP substrate concentration does not prevent inhibition of hydrolysis by MEDI9447, indicating the mAb acts as a non-competitive inhibitor. (B) In contrast, the inhibitory activity of APCP can be overcome by increasing the concentration of substrate to outcompete APCP for binding the active site. Error bars represent standard deviation (SD) from triplicate measurements.
MEDI9447 epitope and paratope mapping
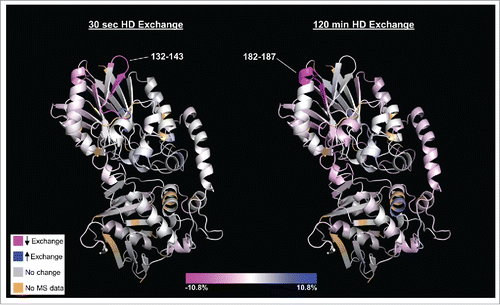
The non-competitive activity of MEDI9447 suggests an allosteric mechanism whereby the mAb binding induces a structural change in CD73 that prevents AMP hydrolysis without affecting substrate binding. Hydrogen deuterium exchange (HDX) mass spectrometry has proven to be a powerful tool to map sites of protein-protein interaction and to characterize protein structure and conformational dynamics.Citation38-40 In order to identify the binding interface of MEDI9447 and CD73, and to test whether MEDI9447 binding affects the conformation of CD73, we performed HDX analysis of MEDI9447 antigen-binding fragment (Fab) and sCD73 either alone or in complex. HDX data were measured from 139 peptides, covering 97.5% of the CD73 protein (standard deviation of ±0 .6 daltons for each exchange time point; ±1 .6 daltons for sum of all 7 time points). Comparing the kinetics of exchange between free and complexed CD73 revealed 2 regions located within the N-terminal domain of sCD73 (amino acids (aa) 132–143 and 182–187) that exhibit decreased deuterium uptake when bound to MEDI9447 Fab ( and Fig. S1A,B). Region 132–143 showed an ˜6% decrease in uptake at the shortest exposure time point, but negligible difference at the longest time point (<0 .5%). In contrast, the degree of differential uptake of deuterium in region 182–187 increased with exposure time (˜3.5% difference at 30 second time point versus ˜10.8% at 2 hours). These results suggest that, of the 2 regions, 182–187 may be the primary site for antibody interaction. (). Analysis of the MEDI9447 Fab showed the complementary-determining regions (CDRs) 1 and 3 of the heavy chain, and CDR1 and CDR2 of the light chain displayed the greatest decrease in exchange when in complex with CD73, suggesting that these regions are the primary constituents of the paratope (Fig. S1C,D). Although CD73 aa 132–143 and 182–187 are discontinuous in the amino acid sequence, they are spatially adjacent when mapped onto the folded structure of CD73 () and are within the N-terminal catalytic domain of the enzyme. Differences in hydrogen exchange were observed at other regions within CD73; however, the majority of mass changes, including those of peptides containing known substrate-binding and active site residues, were not statistically significant (Fig. S1E). These results suggest that MEDI9447 binds to a discontinuous epitope on CD73, and that binding does not alter the conformation of the catalytic domain.
Figure 2. HDX heat map depicting those regions of CD73 (PDB code 4h2f) that undergo differential deuterium uptake when bound to MEDI9447. Relative exchange between antibody-bound and unbound CD73 is depicted as a function of deuterium exposure time with decreased exchange in magenta, increased exchange in blue, and no change in white. The N-terminal region at positions 132–143 exhibits the greatest decrease in exchange at the shortest exchange time point (left image, 30 sec) whereas region 182–187 exhibits the highest degree of differential exchange at the longest time point (right image, 120 min). Regions colored in light brown correspond to residues not detected in the mass spectrometry analysis (2.5% of total sequence). The orientation is such that the N-terminus is at the top and C-terminus is at the bottom of the structure. Color scale bar represents relative % change in deuterium uptake by CD73 in MEDI9447-bound vs. unbound states.
To validate and further refine the interface defined by HDX, we performed epitope mapping using a domain-swapping mutagenesis approach that we have previously described.Citation41,42 As a domain-swapping template, we used the Gallus gallus (chicken) homolog of CD73, which shares ˜65% sequence identity with mature human CD73 but is not recognized by MEDI9447 (Fig. S2). Binding analysis of MEDI9447 to chimeric human-chicken CD73 protein constructs was performed in order to identify those regions required for interaction. Exchanging the protein regions at aa 132–143 and 182–187 between chicken and human CD73 decreased, but did not abolish binding (Table S1). This finding indicates that additional residues outside the HDX-identified interface compose the epitope.
To fully define the MEDI9447 binding site, we generated chimeric CD73 constructs with swapped sequences spanning the entire length of the protein, as well as point and combinatorial mutations (Table S1). Measuring MEDI9447 binding to this panel of human CD73 protein knock-out variants revealed that V144, K180, and N185 are the primary epitope residues, with N185 being the most critical (). Mutating K180A and V144K together results in a further reduction in binding, whereas combining the N185G mutation with either K180A or V144K ablates binding (). In addition to K180, we found Y135, K136, and N187, 3 residues conserved in human and chicken CD73, contribute to MEDI9447 binding, but to a lesser extent ( and Supplementary Table 1). Interestingly, all 4 amino acids were contained in the HDX defined epitope, and conservation between chicken and human CD73 would not point to these residues as being critical for binding. However, the effect of these latter 3 residues was revealed by mutating them to alanine in the context of a domain-swapped background; as exclusive point mutations they have minimal or no measurable effect on affinity (Supplementary Table 1). To confirm V144, K180, and N185 are critical constituents of the epitope, we knocked in V144 and N185 to the corresponding positions in chicken CD73. Encoding only these 3 residues conferred MEDI9447 binding at sub-nanomolar affinity (KD = 79 pM) () within fold10- of the mAb affinity to wild type human CD73, demonstrating that binding is primarily mediated by these 3 amino acid positions. Although these findings show that the HDX analysis identified the general location of the binding interface, 2 of the 3 critical epitope residues (V140 and K180) were not contained within peptides that exhibited differential hydrogen exchange ().
Figure 3. The MEDI9447 epitope resides within the N-terminal domain of CD73. Wild-type (A) and knock-out mutant CD73 proteins (B-F) were immobilized via their HisCitation6 tag on a HTG sensor chip and then binding of MEDI9447 dilutions (5 nM to 0.3 nM, except for (E) at 20 nM to 1.25 nM) was measured by SPR. The mutations V144K (B), K180A (C), N185G (D), and V144K+K180A (E), all reduce MEDI9447 binding. Combining N185G together with either V144K (F) or K180 (data not shown) abolishes binding. (G) SPR kinetics of MEDI9447 binding to wild-type and mutant CD73 proteins. *2:1 fit (see Methods).
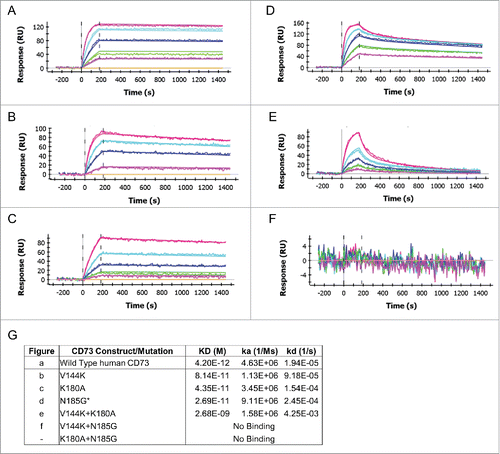
Figure 4. The MEDI9447 epitope is positioned at the apex of the N-terminal domain. (A) Evaluation of MEDI9447 binding to a panel of CD73 knockout and knock-in variants (see Fig. S2 and Supplementary Table 1) revealed 6 residues that constitute the interaction site. Two of the 3 most impactful residues (magenta) are located outside the HDX interface regions (gray). Three less crucial residues (pink) are located within the HDX interface. (AA, amino acid). (B) Knocking in N185 and V144 (K180 is conserved) to a CD73 construct encoding chicken N- and C-terminal domain sequence confers binding to within fold20- the KD for wild-type human CD73 (MEDI9447 dilutions from 5 nM to 0.3 nM; compare to ). (C) Crystal structure of the open conformation of CD73 showing the position of the epitope at the apical, lateral surface of the N-terminal domain. The CD73 N-terminal domain, linker region, and C-terminal domain are depicted in yellow, orange, and blue, respectively. Residues mediating binding are colored as in (A) and shown in blowout, top view. The location of the MEDI9447 epitope is distant from the substrate binding site (adenosine depicted in spheres, C-terminal domain) and the zinc ion coordination site (side chains in cyan, N-terminal domain).
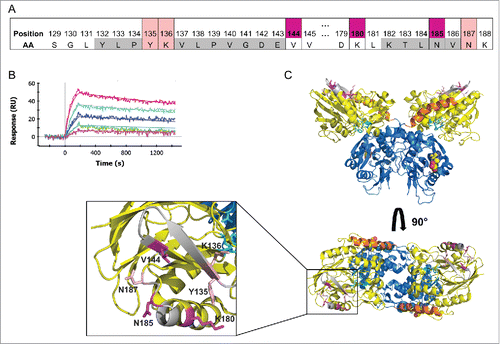
Overlaying the identified epitope onto the structure of CD73 shows that the binding site is located at the apical, lateral surface of CD73 in the open conformation (). Residue N185 is positioned near the N-terminal domain apex in a loop region extending outward from helix G, which also contains the critical residue K180 (). The conserved residues Y135 and K136 are located on β-strand 6 adjacent to K180, while V144 is positioned within β-strand 7, proximal to N187 (). Within the context of the CD73 monomer, the epitope is both on the opposing face and spatially distant from the substrate binding site (). Additionally, the binding site does not encompass any active site residues, including those that coordinate interaction with Zn2+ co-factor (). Thus, the position of the epitope is consistent with the observation that MEDI9447 does not compete for AMP binding; however, it is not apparent how the mAb inhibits CD73 enzymatic activity.
MEDI9447 prevents the conformational transition of CD73 to the active state
Previous structural studies of CD73 demonstrated that the enzymatic activity of CD73 requires a transition between an open and closed conformation states that occurs through extensive N-terminal domain rotation.Citation23 Based on the location of the epitope, we hypothesized that each Fab arm of MEDI9447 IgG engages with separate CD73 N-terminal domains to potentially form a bridge between adjacent membrane-bound or soluble CD73 molecules, and restricts the transition of the enzyme from the inactive, open state, to the catalytically active, closed state. In this proposed model, both Fab arms of the IgG must interact with CD73. To test the requirement of engaging each Fab arm, we measured inhibition of sCD73 as a function of increasing concentrations of MEDI9447 in either an IgG or Fab format. MEDI9447 IgG inhibited sCD73 activity in a dose-dependent manner, with maximal inhibition achieved at a molar ratio of ˜1:1 between the IgG and sCD73 dimer. However, when IgG was in stoichiometric excess relative to sCD73, a loss of inhibition was observed (). This so-called “hook effect” has been observed in other immunoassays and can result from monovalent antibody binding driven by Fab arms on the same IgG molecule competing for limiting binding sites on the target antigen.Citation43 Consistent with this observation, MEDI9447 Fab did not inhibit sCD73 activity (). Together these results indicate that a bivalent interaction of MEDI9447 is required to inhibit sCD73 function.
Figure 5. MEDI9447 inhibits the transition of sCD73 to the conformationally active structure. (A) Dose response of MEDI9447 IgG, Fab, or control IgG on the inhibition of sCD73 hydrolysis of AMP as measured using the CellTiterGlo assay. MEDI9447 IgG reaches maximal inhibition at a 1:1 molar stoichiometry with CD73 dimer (arrow). At high concentrations, where MEDI9447 IgG is in excess(>10 nM), a loss of inhibition or “hook effect” is observed. MEDI9447 Fab and control IgG do not inhibit sCD73. Assay was performed using the CellTiterGlo assay as described in Methods (RLU, relative light units). (B) Wild type CD73 was immobilized on a HIS2 biosensor and binding of MEDI9447 (blue sensorgram) and anti-CD73 mAb A (brown sensorgram) was measured by BLI. CD73 pre-incubated with Zn2+ and APCP retains binding to MEDI9447 (black sensorgram) but mAb A binding is lost (orange sensorgram). (C) Although Zn2+ and APCP pre-incubation with CD73 cause a loss in mAb A binding (orange sensorgram), pre-incubation with MEDI9447 before addition of Zn2+ and APCP restores binding (black sensorgram). Binding of mAb A to CD73 alone, CD73 pre-incubated with MEDI9447 (but not Zn2+ and APCP), and MEDI9447 is shown in the brown, blue, and purple sensorgrams, respectively. (D) Cartoon model depicting how MEDI9447 (not depicted) may prevent CD73 from adopting the fully closed, active conformation induced by Zn2+ and APCP; mAb A (red) only binds to CD73 in the open and “intermediate” conformation. Error bars (A), and dots (B,C) represent SD.
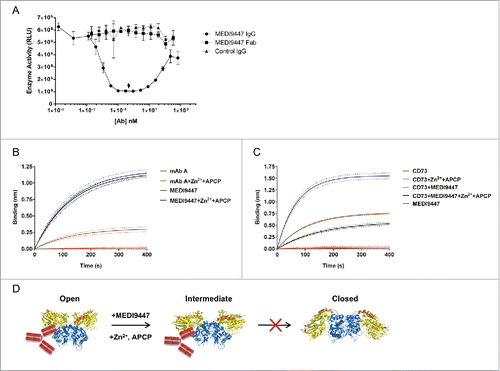
To examine whether MEDI9447 IgG inhibits CD73 conformational transition, we employed an anti-CD73 mAb, mAb A, as a reporter of CD73 conformation. Mapping the binding interface of mAb A with CD73 revealed an epitope distinct from MEDI9447 that interacts with both the N- and C-terminal domains (Fig. S3A-D). Considering the location of the mAb A binding region, we postulated that the epitope would be present in the CD73 open conformer, but disrupted in the closed conformer (Fig. S3B). To confirm this, we measured binding of mAb A to open, substrate-free, sCD73 and closed sCD73 (). To induce the closed conformation, we pre-incubated sCD73 with Zn2+ and APCP, the co-factor and the non-hydrolyzable substrate previously used to generate the crystal structure of the closed conformer of human CD73.Citation21 Analysis using biolayer interferometry (BLI) demonstrated mAb A binds to the open, inactive sCD73; however, binding is abolished when CD73 is pre-incubated with ZnCitation2+ and APCP to induce the closed conformer (). This finding is consistent with the mAb A epitope being present in only the open structure of CD73. In contrast, MEDI9447 binding was not sensitive to the state of CD73, indicating that it can bind to either substrate-free or bound CD73 (). Having established that mAb A binding reports on the conformational state of CD73, we investigated what effect MEDI9447 may have on the ZnCitation2+/APCP-induced structural transition of CD73 to the active state. mAb A bound to sCD73 that was pre-incubated with MEDI9447, confirming that the 2 antibodies bind to distinct epitopes (). Notably, when the sCD73-MEDI9447 pre-complex was subsequently incubated with Zn2+ and APCP, binding by mAb A was maintained (), suggesting CD73 was in the open state, even in the presence of substrate and cofactor. In contrast, a control IgG and MEDI9447 Fab did not restore mAb A binding when pre-complexed with sCD73 prior to Zn2+ and APCP addition (Fig. S4). These results support the hypothesis that bivalent MEDI9447 binding prevents the transition of sCD73 from the open state to the fully closed, hydrolytically active conformation ().
Formation of inter-dimer bridged complexes of CD73 with MEDI9447
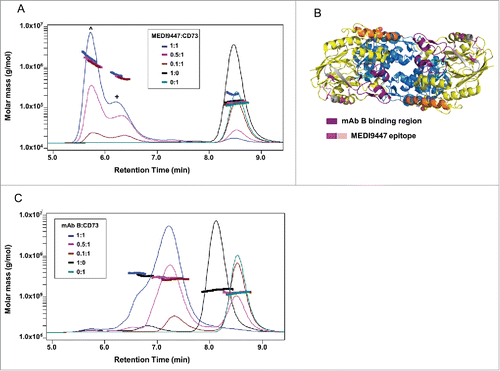
The observed inhibitory activity of MEDI9447 could occur either through bridging of N-terminal domains belonging to separate CD73 dimers (inter-dimer bridge) or a single CD73 dimer (intra-dimer bridge). To discriminate between these scenarios, we characterized the size of the complexes formed in solution. Based on the measured mass of unbound MEDI9447 and sCD73 (˜145 and ˜125 kDa, respectively), the predicted size of an intra-dimer bridged 1:1 complex of antibody to sCD73 would be ˜270 kD (Fig. S5A). We employed size-exclusion chromatography - multi-angle light scattering (SEC-MALS) to predict the molecular mass shift of the MEDI9447 complexed with sCD73. When MEDI9447 and sCD73 were incubated at a 1:1 molar ratio, 2 complexes were formed. The predominant species had a weight average molar mass (Mw) of ˜1.74 megadaltons and the less abundant species had a Mw of ˜0.66 megadaltons (). The Mw of the largest complex is consistent with an oligomer containing 7 CD73 dimers and 6 IgGs (7 × 125 kDa + 6 × 150 kDa = 1.745 megadaltons). When MEDI9447 is in limiting concentrations (0.5:1, 0.1:1 mAb:antigen), complexes of comparable Mw are formed, but the relative difference in abundance of each species is less pronounced (). To determine whether oligomer formation is a common effect of any mAb targeting the N-terminal domain of CD73, we characterized complexes formed with a different anti-CD73 antibody, mAb B. mAb B binds to a region within the N-terminal domain of CD73 that, in contrast to MEDI9447, is proximal to the groove formed between the CD73 monomers ( and Fig. S5B,C). Unlike MEDI9447, mAb B forms complexes of ˜270–295 kDa, a Mw close to that predicted for a 1:1 interaction (). Since CD73 is found both as soluble and membrane-bound forms, we investigated whether MEDI9447 can bridge native, endogenous CD73 dimers anchored to the cell surface. Using western blot analysis, we compared the size of complexes formed when CD73-expressing cells were incubated with either MEDI9447 or control antibodies. MEDI9447 IgG, but not Fab, mAb B, nor a control IgG, formed oligomeric complexes when bound to cell-surface CD73 (Fig. S5D). Collectively, these findings indicate that MEDI9447 forms inter-dimer bridges between multiple CD73 dimers, and the generation of these oligomers is conferred by the epitope.
Figure 6. MEDI9447 forms inter-dimer bridges between soluble CD73 molecules. CD73 was incubated with varying amounts of MEDI9447 or anti-CD73 mAb B and analyzed by SEC-MALS. Shown in (A,B) are SEC UV chromatograms with protein retention time on the x-axis and molar mass distributions determined by MALS indicated across the elution profiles with the corresponding mass on the y-axis. (A) At a 1:1 molar ratio (blue trace), MEDI9447 forms complexes with CD73 of ∼1.7 (ˆ) and ∼0.66 (+) megadaltons. Comparably sized complexes are formed at lower ratios of MEDI9447:CD73 (0.5:1 in magenta, 0.1:1 in brown). MEDI9447 and CD73 alone are represented by the black and teal UV traces, respectively. (B) Top-down view of the crystal structure of CD73 dimer showing the mAb B binding region (purple) and MEDI9447 epitope (magenta and pink). (C) CD73 bound to mAb B forms a single predominant complex of ∼270–290 kD (peak at ∼7.2 min). UV traces shown represent 1:1 mAb B:CD73 (blue), 0.5:1 (magenta), and 0.1:1 (brown). mAb A and CD73 alone are in black and teal, respectively.
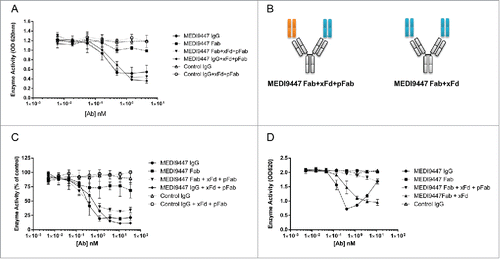
MEDI9447 inhibits anchored CD73 via monovalent interaction
Although CD73 is shed from the cell surface in vivo and retains enzymatic activity in its soluble form, the majority of native CD73 exists in a GPI-anchored format.Citation44,45 In our preceding studies we found that MEDI9447 IgG, but not Fab, inhibits sCD73. To interrogate whether MEDI9447 also inhibits the activity of immobilized CD73, we used a defined assay system whereby recombinant sCD73 is captured via a C-terminal 6 histidine tag to confer a spatial orientation resembling that of GPI-anchored CD73. Similar to our previous results, MEDI9447 IgG inhibited AMP hydrolysis in a dose-dependent manner (). However, when the antibody was in molar excess relative to CD73 dimer, no loss of inhibition, or hook effect, was observed ( and Fig. S6). Unexpectedly, MEDI9447 Fab also inhibited CD73 activity with similar potency but a lower maximal inhibition compared to MEDI9447 IgG ( and Fig. S6). These results indicate that, unlike soluble CD73, immobilized CD73 can be inhibited via monovalent interaction with MEDI9447. The difference in inhibition between the IgG and Fab suggested that the size of the antibody molecule may dictate the level of enzyme inhibition. To investigate this, we pre-incubated MEDI9447 Fab with an anti-Fd antibody (xFd) under conditions such that one Fab arm of the xFd antibody is bound to a MEDI9447 Fab and the other arm is bound to a non-specific, polyclonal Fab (pFab) (). Formation of this complex effectively increases the size of MEDI9447 Fab while maintaining monovalent binding to CD73. This xFd-captured Fab inhibited immobilized CD73 activity to an equivalent degree as MEDI9447 IgG (). To extend this observation to native, GPI-anchored CD73, we measured antibody inhibition of endogenously expressed CD73 in the human epithelial breast cancer cell line, MDA-MB-231.Citation46 Similar to immobilized recombinant CD73, GPI-anchored CD73 was inhibited robustly by MEDI9447 IgG, modestly by MEDI9447 Fab, and pre-binding Fab to the xFd antibody increased maximal inhibition to a level equivalent to IgG (). Lastly, we tested inhibition of non-immobilized sCD73 by MEDI9447 Fab bound to either one or both arms of the xFd antibody. Unlike cell-surface CD73, sCD73 was not inhibited by MEDI9447 Fab bound to a single xFd arm (). However, conferring bivalency by binding MEDI9447 Fab to both xFd arms resulted in sCD73 inhibition comparable with MEDI9447 IgG (). These findings show that surface-anchored CD73 can be inhibited by monovalent antibody binding and that efficacy is mediated by the size of the antibody. This is in direct contrast to soluble CD73, which is only inhibited by MEDI9447 through bivalent interaction.
Figure 7. Surface-bound CD73 is inhibited by both IgG and Fab formats of MEDI9447. (A) CD73 was immobilized via a C-terminal histidine tag to a nickel-coated microtiter plate and inhibition of AMP hydrolysis by MEDI9447 IgG, Fab or control antibodies was measured using the Malachite Green assay. MEDI9447 IgG, but not control IgG, inhibits immobilized CD73 hydrolysis of AMP in a dose-dependent manner. MEDI9447 Fab also inhibits CD73 activity, but to a lower extent. (B) When MEDI9447 Fab (cyan) is bound to one arm of an anti-Fd antibody (xFd, gray) and the other arm bound to a non-specific polyclonal Fab (pFab, orange, (B), left cartoon) inhibition is increased to that comparable with MEDI9447 IgG through increasing the effective size rather than valency. (C) Enzyme activity of endogenously expressed CD73 in MDA-MB-231 cells measured by CellTiterGlo assay. As with immobilized recombinant CD73, MEDI9447 IgG inhibits AMP hydrolysis to a greater degree than the Fab, and increasing the effective size of the MEDI9447 Fab by complexing with an anti-Fd antibody enhances inhibition. (D) To test whether the xFd+MEDI9447 can inhibit soluble CD73, AMP hydrolysis was measured using the Malachite Green assay. MEDI9447 Fab either alone or bound to a single xFd arm does not inhibit soluble CD73 activity. In contrast, MEDI9447 Fab binding to both xFd arms (MEDI9447 Fab + xFd, (B), right cartoon) confers bivalency, resulting in CD73 inhibition. Error bars represent SD.
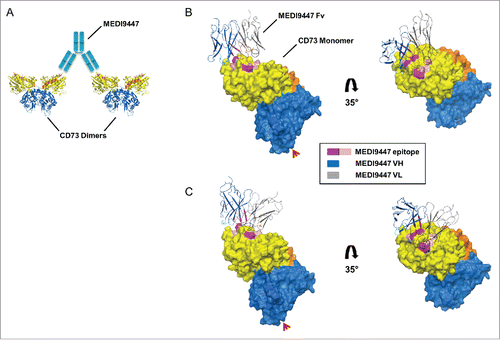
Docking of MEDI9447 to CD73
Bridging separate CD73 dimers by MEDI9447 would require each bound Fab arm to be oriented outward from the dimer interface to permit the opposing Fab arm to extend and bind to the neighboring CD73 dimer. This bridging configuration is most feasible if the antibody heavy chain variable domain (VH) and light chain variable domain (VL) interact with the lateral and medial edges of the CD73 N-terminal domain, respectively (). To investigate the potential geometry of bound MEDI9447, we first built a homology model of the MEDI9447 variable fragment (Fv) and then used the model to perform computational protein-protein docking analysis to the human CD73 crystal structure. Cluster analysis of 70,000 pose rotations revealed several pose clusters with MEDI9447 binding to the epitope surface in orientations consistent with CD73 bridging (). The VH CDRs are positioned at the lateral edge of CD73, though the projection angle of the Fv varies depending on the particular cluster pose ( vs C). Thus, although a co-crystal structure is necessary for absolute determination of the binding mode, in silico docking analysis predicts multiple binding configurations that are consistent with inter-dimer bridging.
Figure 8. Computationally predicted models of MEDI9447 Fv bound to CD73. (A) Cartoon depiction of how CD73 inter-dimer bridging would be facilitated by the MEDI9447 IgG (cyan) VH and VL interacting with the lateral and medial edges of the binding site, respectively. (C,B) Pose cluster examples showing the Fv model structure extending outwards from the CD73 monomer, though at different projection angles relative to the binding site (CDR H1, H2, H3 in cyan, teal, and blue, and CDR L1, L2, L3 in black, orange, and brown, respectively; Chothia annotation). Color coding of the CD73 structure is as in . The Fv VH and VL are represented in gray and dark blue, respectively. Red arrowheads indicate the GPI anchor attachment site.
Discussion
In this study, we report the epitope and mechanism of action of MEDI9447, a therapeutic mAb that inhibits the enzymatic activity of CD73. Our results reveal a binding site within the CD73 N-terminal domain that enables inhibition through 2 distinct, conformation-mediated mechanisms. Importantly, this feature confers MEDI9447 the ability to block both soluble and cell surface anchored CD73 in a non-competitive manner.
An ideal therapeutic molecule targeting CD73 would be specific in its neutralizing action to avoid affecting the substrate interaction of structurally related nucleotidases or nucleotide/side binding proteins. In this regard, selective inhibitors that act allosterically or through other non-competitive mechanisms may be preferred, both in terms of avoiding off-target activity and the need to compete with multiple endogenous substrates that also bind to the enzyme active site.Citation47,48 Our finding that MEDI9447 does not block CD73 substrate binding renders it an attractive therapeutic; this mode of inhibition confers specificity for CD73 unlike existing, non-selective small molecule inhibitors that target the structurally conserved active site.Citation28,49 Additionally, MEDI9447 is not expected to compete in vivo with active-site binding CD73 substrates, an attribute that might translate to greater potency. Our HDX and mutagenesis studies revealed that MEDI9447 binds a discontinuous epitope within the CD73 N-terminal domain. Absent a co-crystal structure, we cannot formally exclude the possibility that some CD73 residues that we discounted as interactors with MEDI9447 do indeed form contacts with the paratope. However, our extensive CD73 mutant binding analysis indicates that these potential residues would contribute minimally to the thermodynamics of interaction. Given the importance of heavy chain CDR3 in forming antigen contact sites, it is not unexpected that this CDR exhibited the greatest degree of differential hydrogen exchange. The HDX results also suggest that the light chain contributes to the paratope, particularly residues within CDR1 and 2.
The location of the epitope at a position distal from the CD73 substrate binding site is consistent with MEDI9447 having a non-competitive mode of inhibition. Based solely on the binding interface, one might hypothesize that the antibody acts as a classic allosteric inhibitor, inducing a long-range conformational change in CD73 that distorts active site residues in a manner that prevents hydrolytic activity. However, our HDX data did not reveal significant alterations in CD73 structure in regions outside the binding site that would support this form of allostery. Alternatively, the epitope could suggest that MEDI9447 binding restricts movement of loops, β-strands, or α helices within the N-terminal domain that is required for catalytic activity. Conversely, little to no difference in secondary or tertiary structure within the N-terminal domain was previously reported between the open and closed structures of CD73; the local, inter-domain conformational changes that do occur are restricted to the linker and C-terminal domain.Citation21
We propose that the location of the MEDI9447 epitope enables antibody bridging or cross-linking of CD73 dimers in a manner that restricts the necessary conformational change, rather than inducing a structural change. Our data demonstrating that bivalent IgG is required to inhibit the soluble form of CD73 and the mAb forms complexes containing multiple CD73 dimers supports the mechanism of MEDI9447 bridging of CD73 molecules. This latter result is in contrast to that of mAb B, which also binds the N-terminal domain but does not form inter-dimer bridges, highlighting the importance of the MEDI9447 epitope in conferring this cross-linking activity. To achieve bridging, it is expected that each bound Fab arm of MEDI9447 projects outward, away from the CD73 dimer interface, a geometry that is supported by several computationally predicted structures of the complex. Although future crystal structure studies are required to reveal the exact binding interaction, our functional studies suggest that the binding configuration likely will be similar to the in silico models. Rationally, if the N-terminal domains of 2 separate CD73 dimers are bound by Fab domains of the same IgG, this could physically restrict the movement of the N-terminal domain and linker region that is required for the enzyme to adopt its closed, active structure. Through the use of mAb A, which serves as a conformational probe of CD73 state, we demonstrated that binding of MEDI9447 inhibits CD73 from adopting the fully closed conformer, induced by Zn2+ and APCP. The intermediate level of binding by the reporter mAb A when MEDI9447 was pre-complexed with CD73 suggests that some degree of conformational transition is still induced by Zn2+ and APCP. This diminished mAb A binding may reflect that its epitope is partially distorted in this inactive intermediate state, but still sufficient to allow binding. Given the high degree of flexibility of both the CD73 and the IgG hinge regions, and because MEDI9447 is a non-competitive inhibitor, it is not surprising that even when the mAb bridges 2 CD73 dimers, there is still some CD73 structural alteration due to substrate binding. In this mode of action, MEDI9447 can inhibit the activity of both soluble and surface-bound CD73.
More remarkably, while interrogating the binding epitope of MEDI9447 and discerning how this unique epitope could in fact inhibit enzymatic activity through bivalent bridging of CD73, we discovered an additional mode of action: MEDI9447 can also inhibit surface-bound CD73 through a monovalent binding mechanism. Although initially perplexing, a second, steric-mediated blocking mechanism of anchored CD73 activity is consistent with our observations. The formation of catalytically active, GPI-anchored CD73 requires that the N-terminal domain rotate downwards, to a position proximal to the cell surface.Citation21 The native CD73 dimer is ˜130 kDa.Citation50 Compared to the size of an IgG or Fab (˜150 kDa and ˜50 kDa, respectively), it is not surprising that antibody bound to the N-terminal domain could sterically block CD73 from fully rotating to adopt the closed conformation. This mechanism is supported by 2 observations. Firstly, MEDI9447 Fab exhibits a lower maximal inhibition compared to the IgG, which is consistent with a size-dependent steric effect. The disparity between Fab and IgG is unlikely due to differences in binding kinetics since the Fab binds CD73 with sub-nanomolar affinity (KD= 327 pM, data not shown). Further, the elevated activity conferred by increasing the effective size of the Fab through conjugation with an anti-Fd antibody also strongly supports a size, and not a valency or avidity-dependent, steric mechanism. The second observation supporting this mode of inhibition is that a hook effect, or loss of inhibition, is not observed with MEDI9447 IgG when CD73 is surface bound. Presumably, this is because the antibody can block anchored CD73 hydrolysis of AMP through either a bi- or monovalent interaction, regardless of CD73 surface density or antibody concentrations. In contrast, a hook effect is observed with soluble CD73 due to the absence of an anchoring or solid-phase capture surface required for the monovalent Fab or a molar excess of IgG (monovalent IgG binding) to sterically block the N-terminal domain rotation. Under the tested conditions, this hook effect was observed with MEDI9447 IgG, but not MEDI9447 Fab captured on both arms of an anti-Fd antibody (). However, the anti-Fd/Fab complex exhibited lower inhibitory activity, possibly reflecting decreased CD73 binding activity, which suggests a higher concentration of the anti-Fd/Fab complex is required to reach the hook effect threshold.
In vitro, we found that inhibition of sCD73 by MEDI9447 was maximal at a ˜1:1 molar ratio (). Whether patients receiving MEDI9447 would achieve this molar ratio is unknown at this time, and may vary depending on the patient, antibody pharmacokinetics, tumor type, and compartment (e.g., tumor vs serum). However, as soluble CD73 is a minor fraction of the total system CD73, it would not necessarily be optimal to dose MEDI9447 with the goal of achieving a 1:1 ratio with sCD73. Rather, a clinical study should be designed to ensure sufficient suppression of CD73 to prevent adenosine production and signaling in the tumor microenvironment, as opposed to inhibiting sCD73 in the periphery.
We propose a model whereby MEDI9447 antagonizes soluble and GPI-anchored CD73 function through a dual mechanism of inhibition that is integrally linked to its epitope (Fig. S7). Although our studies show that MEDI9447 can block both soluble and bound CD73, it is unknown whether GPI-anchored CD73 would be inhibited preferentially through one mechanism in the in vivo setting. Presumably, the density, orientation, and inter-dimer distance of CD73 on the cell surface would dictate the dominant mode of inhibition. Given that many cancer cells overexpress CD73, which would increase the likelihood of the dimers being in close proximity, it is expected that MEDI9447 would engage in both bi- and monovalent interactions. On cells with relatively low CD73 expression, MEDI9447 might inhibit AMP hydrolysis primarily through the steric blocking mode. Postulating beyond this specific interaction with CD73, the dual mechanism of MEDI9447 exemplifies how antibody therapeutics can be employed to restrict the catalytic domain formation as a strategy that could be applied to other ecto-enzymes that play numerous roles in both normal and disease biology.Citation51,52
From a mechanistic perspective, blocking CD73 is an immuno-oncology strategy distinct from that of related targets such as PD-1 and CTLA-4, for which there are already approved drugs. From a therapeutic standpoint, the activity of MEDI9447 is advantageous. It non-competitively inhibits CD73, and, therefore, does not compete with endogenous nucleotide binding to the active site. This avoids any potential cross-reactivity toward other nucleotide/side binding proteins with structurally conserved catalytic sites, and importantly, does not require the blocking of multiple substrates at the active site. Further, MEDI9447 can inhibit both soluble and membrane-bound CD73 through either mono- or bivalent engagement. Both of these features would be expected to contribute to in vivo efficacy. The immune-modulating potential of MEDI9447 is currently being evaluated in the clinic for the treatment of cancer, both alone and in combination with existing therapeutic agents that target complementary immune modulating pathways. Understanding the mechanism of action of MEDI9447 through the dual mode of activity provides insights into the first therapeutic targeting CD73 in the clinic.
Materials and methods
CD73 and antibody reagents
The mammalian expression plasmid encoding recombinant mature human CD73 (amino acid positions 1–526) was constructed at MedImmune. To achieve expression of a soluble, secreted form of CD73, the GPI anchor signal peptide was removed and replaced with a C-terminal 6x-histidine tag. CD73 sequence numbering is based on the mature protein without the signal peptide (human NT5E, NCBI reference sequence NP_002517.1). Plasmids encoding the recombinant human/chicken chimeric domain swapped “knockout” (KO) mutants were generated using synthetic DNA gBlocks (IDT, Inc..) encoding codon optimized chicken CD73 DNA sequence (chicken NT5E, NCBI reference sequence XP_004940453.1). Based on alignment with human CD73 protein sequence, amino acid coding position 1 of constructs containing a chicken N-terminal domain corresponds to position 20 of immature chicken CD73 containing the predicted signal peptide. Full-length KO DNA constructs were made by single overlap extension PCR of gBlocks and PCR amplicons of human CD73. All constructs contained a C-terminal 6x histidine tag. Single and multiple point mutations in human and chimeric constructs were made by site-directed mutagenesis using the Quick Change Lightning Multi Site-Directed Mutagenesis Kit (Stratagene). All CD73 constructs were expressed in suspension HEK293 cells. Histidine-tagged wild-type human CD73 (lacking the GPI anchor signal sequence) was purified in-house using a HisTrap nickel affinity column (GE Healthcare Life Sciences). The protein was confirmed to be a dimer in solution, with a molar mass of ˜125 kDa (see Fig. S5A). All mutant CD73 constructs were expressed by transiently transfecting suspension HEK293 cells using 293Fectin (Life Technologies). Cells were grown and transfected in serum-free 293Freestyle media (Life Technologies) in 24-well, deep-well blocks. Crude cell supernatants were harvested 6 d post-transfection and filtered through a 0.45 μm filter to remove cell debris before use. Supernatant concentrations of CD73 variants were determined by measuring binding of the histidine tagged proteins to HIS2 biosensors (FortéBio/Pall Life Sciences) on an Octet QK384 bio-layer interferometry (BLI) instrument (FortéBio/Pall Life Sciences). Concentrations were calculated using the Octet data analysis software by comparing binding signal to a standard curve generated from dilutions of purified, recombinant 6x-histidine tagged human CD73 of a known concentration. MEDI9447, mAb A, and mAb B were discovered by phage display. V-genes were isolated from DP47 scFv phage library by phage display panning on recombinant CD73 protein.Citation53 Primary screening was performed in scFv-Fc using binding assays to recombinant antigen and cell lines, CD73 enzymatic activity inhibition on recombinant proteins and cell lines, and internalization assays (data not shown).Citation54 MEDI9447 (human IgG1-TM and mouse IgG1 formats),Citation55 mAb A (human IgG1), and mAb B (human IgG1) and MEDI9447 Fab (human IgG1) were expressed and purified in-house. IgGs were expressed in mammalian cells and purified by Protein A and size-exclusion chromatography (HiTrap Protein A and Superdex 200, GE Healthcare Life Sciences). To generate MEDI9447 Fab, 10 mgs of IgG were digested for 5 hr at 37°C with immobilized papain (Thermo Scientific/Life Technologies) and the Fab was purified using a HiTrap Q column (GE Healthcare Life Sciences).
HDX analysis
Recombinant human CD73 and MEDI9447 Fab samples were prepared at a concentration of 2 mg/mL. The CD73 + Fab complex was formed by pre-incubation at a mass ratio of 1:1(mg:mg). The entire HDX experiments were carried out using Waters HDX Technology (Waters Corporation) equipped with a Leap automation robot. Briefly, 1.25 µL protein samples were diluted 20 times with either H2O buffer (10 mM phosphate, pH 7.0) or D2O buffer (10 mM phosphate, pD 7.0) at 20°C. After incubation at various time points (0 second for undeuterated experiments or 0.5, 1, 5, 10, 30, 60, and 120 minutes for deuterated experiments), the labeled samples were quenched by adding an equal volume of an ice-cold solution of 4.0 M guanidine HCl (Pierce Biotechnology), 500 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (Pierce Biotechnology), pH 2.4. Immediately thereafter, samples were digested using a Poroszyme Immobilized Pepsin cartridge (Applied Biosystems) at 20°C. The peptic fragments were collected and desalted using a ACQUITY BEH C18 VanGuard Pre-column (2.1×5 mm, Waters), and eluted into an ACQUITY BEH C18 column (1.7 μm, 1.0 × 100 mm, Waters) at 0°C. Peptides separated on the column were analyzed by a SYNAPT G2 mass spectrometer (Waters). For data analysis, peptides were identified using ProteinLynx Global Server software (Waters), and the deuterium incorporation levels for each peptic peptide from each labeling time point were calculated using DynamX (Waters) followed by manual datapoint inspection. For each protein, 4 undeuterated experiments and 3 complete HDX experiments were performed. The significant difference values (standard deviation in daltons) were calculated using the experimental uncertainty and a 98% confidence interval as previously described, except that a pooled variance of standard deviations was used instead of the mean of individual standard deviations.Citation56 The relative fractional uptakes between the CD73 + Fab complex and CD73 were generated by DynamX software (Waters) and exported to PyMOL (Shrödinger, LLC) for structural modeling. All structure figures of human CD73 were generated using PyMOL and the reported crystal structures of the open and closed conformations of CD73 (Protein Data Bank (PDB) code 4h2f and 4h2i, respectively).
SPR and BLI binding analysis
Binding of MEDI9447 to wild-type and mutant CD73 proteins was measured by surface plasmon resonance (SPR) using a ProteOn instrument (BioRad). CD73 crude cell supernatant protein samples were diluted to 3 μg/ml in PBS, 0.005% Tween-20, pH 7.4 (BioRad) and immobilized to ˜400 RU on a HTG tris-NTA sensor chip (BioRad) pre-activated with 10 mM NiSO4, 10 mM MES, pH 6.0 (BioRad). An equivalently diluted crude cell supernatant sample from non-transfected cells was included as a reference channel control. Sensorgrams were recorded by flowing fold2- dilutions of MEDI9447 prepared in PBS, 0.005% Tween-20, pH 7.4, ranging from 5 nM to 0.31 nM. In some cases, for CD73 variants that bound weakly to MEDI9447, antibody dilutions ranged from 20 nM to 1.25 nM. Antibody binding was measured at a flow rate of 100 μL/min, with a 3 min association phase and 20 min dissociation phase. The sensor chip surface was regenerated between each run by an 800 s injection of 300 mM EDTA, pH 8.5 (BioRad) at a flow rate of 30 μL/min. Binding kinetics were analyzed using the ProteOn data analysis software. Double referencing was performed and, unless noted otherwise, a 1:1 Langmuir binding model was utilized to fit the data. Some CD73 variants containing mutations within the amino acid region 171–188 fit poorly to a 1:1 model (i.e., ChiCitation2 values >10% of Rmax). Where noted, these variants were fit with a heterogeneous antigen (2:1) model. Due to the high affinity of MEDI9447 (˜4 pM) and sensitivity limitations of ProteOn, only changes of >2-fold in measured kinetics were considered meaningful when comparing MEDI9447 binding to the CD73 variants. Although the format of the ProteOn assay is not ideal for determining absolute affinity measurements (CD73 immobilized, IgG instead of Fab), it is not anticipated that this impacts ranking of MEDI9447 binding to the different CD73 variants.
Mapping the binding regions of anti-CD73 antibodies mAb A and mAb B was performed by BLI using an Octet QK384 instrument. All proteins were prepared in 1x Kinetics Buffer (FortéBio/Pall Life Sciences). C-terminally histidine tagged CD73 variants from crude cell supernatants were diluted to 6 μg/ml and immobilized on HIS2 biosensors to a binding response threshold of 0.8 nm. After a 300 sec baseline step, sensors were incubated in 30 nM antibody. Association and dissociation times were 600 sec. A non-transfected cell supernatant reference control was included for background binding subtraction during data analysis. Data was processed and graphs prepared using the FortéBio Data Analysis software.
CD73 enzyme activity assays
The CD73-catalyzed hydrolysis of AMP to adenosine and inorganic phosphate was analyzed by either quantifying inorganic phosphate (Malachite Green assay; R&D Systems) or measuring the ATP-dependent oxidation of luciferin which is inhibited by AMP, as previously described (CellTiterGlo assay; Promega).Citation35 Data graphs and enzyme kinetics measurements (Michaelis-Menten non-linear regression) were generated using Prism software (Graphpad). Experiments were performed in either duplicate or triplicate.
For measurements of soluble recombinant CD73 activity using the CellTiterGlo assay, 400 pM recombinant CD73 and various concentrations of anti-CD73 antibodies were incubated in assay buffer (25 mM Tris pH7.5, 5 mM MgCl2, 0.005% Tween-20) for 1 hour at 37°C before adding an equal volume of 200 μM AMP/600 μM ATP (in assay buffer). After 1 hour incubation at 37°C, the AMP concentration in the sample was determined using the CellTiterGlo assay following the manufacturer's instructions.
For measurements of soluble recombinant CD73 activity using the Malachite Green assay,1 nM recombinant CD73 and either 1 nM antibody or 40 μM adenosine 5′-(α,β-methylene)diphosphate (APCP; Sigma-Aldrich) were incubated in assay buffer (25 mM Tris pH 7.5, 5 mM MgCl2, 0.005% Tween-20) for 1 hour at room temperature. An equal volume of 400 μM AMP (for anti-CD73 antibodies) or 3 mM AMP (for APCP) in assay buffer was added and samples were incubated for 15 minutes at room temperature. The concentration of inorganic phosphate was determined using the Malachite Green assay following the manufacturer's instructions.
For measurements of immobilized recombinant CD73 activity, 50 μL CD73 at 100 ng/mL in assay buffer (25 mM Tris pH 7.5, 5 mM MgCl2, 0.005% Tween-20) supplemented with 100 μg/mL BSA was immobilized on nickel-coated plates (Life Technologies). Unbound CD73 was washed away and 50 μL of anti-CD73 antibody (in assay buffer) was added. After incubating for 1 hour at room temperature, plates were again washed, 100 μL of 500 μM AMP (in assay buffer) was added, and samples were incubated for 15 minutes at room temperature. The concentration of inorganic phosphate was determined using the Malachite Green assay following the manufacturer's instructions. In some experiments with both immobilized and soluble CD73, anti-CD73 antibody (IgG and Fab) was pre-incubated for at least 2 hours with fold10- molar excess polyclonal human Fab fragment (Bethyl Laboratories) and 100-fold molar excess sheep-anti-human IgG(Fd) (Meridian Life Sciences) before the addition to CD73.
For measurements of endogenous CD73 activity in cultured cells, 20,000 MDA-MB-231 cells per well were plated in RPMI/10% fetal bovine serum (FBS) (Life Technologies) in a 96-well plate. After an overnight incubation, wells were washed 3 times with serum-free RPMI and 50 μL of antibodies (in serum-free RPMI) were added. After incubating for 30 minutes at 37°C, 25 μL of 1.2 mM AMP (in serum-free RPMI) were added per well. Plates were incubated for 3 hours at 37°C. Twenty-five μL of cell supernatant and 25 μL of 100 mM ATP were mixed and the AMP concentration in the sample was determined using the CellTiterGlo assay following the manufacturer's instructions.
mAb A reporter assay of CD73 conformational transition
Binding of MEDI9447 and mAb A to purified recombinant human CD73 was performed on an Octet QK384 instrument. To compare binding of mAb A and MEDI9447, CD73 was diluted to 6 μg/mL in PBS, pH 7.4 (Life Technologies) and loaded onto HIS2 biosensors to a binding signal threshold of 1.0 nm. The biosensors were then transferred into either PBS alone or PBS with 10 μM ZnCl2 (Sigma-Aldrich), APCP, and/or 2 mM ethylenediaminetetraacetic acid (EDTA) (Life Technologies) and incubated for 15 min. Next, biosensors were transferred to PBScontaining MEDI9447 or mAb A diluted to 30 nM in PBS, and antibody association was measured for 400 sec. Binding signals were referenced to biosensors incubated with CD73 and matching buffer, but not antibody. To test the effect of MEDI9447 on CD73 conformational transition in the presence of ZnCl2 and APCP, mAb A diluted to 10 μg/mL in PBS, pH 7.4 was immobilized on an anti-human Fc AHC biosensor (FortéBio/Pall Life Sciences). Following the 400 s immobilization step, biosensors were blocked for 10 min in non-specific polyclonal human IgG (Jackson ImmunoResearch Laboratories) at 50 μg/mL in PBS, pH 7.4. After a 4 min baseline step, mAb A binding was measured by incubating the biosensors for 400 sec in wells containing CD73 diluted to 250 nM (based on the molecular weight of a dimer) in PBS alone, or PBS containing 10 μM ZnCl2 and/or 0.5 mM APCP. For samples containing MEDI9447 bound to CD73, a mouse IgG1 (used to avoid binding to the anti-human Fc biosensor) version of MEDI9447 diluted to 250 nM in PBS was pre-incubated with the CD73 for 15 min at room temperature before incubation with ZnCl2 and APCP. MEDI9447 Fab and a mouse isotype-matched control IgG1 (generated in-house) were tested at 500 nM. Shake speed for all assay steps was 1000 rpm. Assays were performed independently 3 times. Binding analysis and data graphs were generated using the FortéBio Data Analysis software.
SEC-MALS analysis
For experiments analyzing complexes formed between CD73 and MEDI9447 or mAb B, 900 pmoles of CD73 were incubated with 900, 450, 90, or 0 pmoles of antibody, diluted into PBS, pH 7.4. A separate sample of 900 pmoles of antibody only was also prepared. Samples were incubated for 30 min at room temperature and then 100 μL of each sample was separated on a HP 1100 HPLC (Agilent) using a TSKgel G3000WxL 5 μm, 7.88 mm × 30 cm column (Tosoh Bioscience, LLC) at a flow rate of 1 mL/min for 20 min. Sample running buffer was 0.1 M NaPi, 0.1M NaSO4, pH 6.8. Following HPLC separation, all samples were analyzed using a Dawn Heleos II MALS detector and Optilab T-rEx refractive index detector (Wyatt). Data plots were generated using Astra software (Wyatt). Each experiment was performed on 2 separate occasions; representative data from one experiment are shown.
Cell-surface antibody crosslinking experiment
One million MDA-MB-231 cells were incubated with fold5- serial dilutions of antibody in 1 mL DMEM/10% FBS for 30 min at 4°C. MEDI9447 IgG, MEDI9447 Fab, and control IgG were used at 30 nM starting concentration; mAb B was used at 300 nM starting concentration. Cells were washed in PBS, resuspended in 500 μL BS(PEG)5 (0.75 mM in PBS; LifeTechnologies), and incubated for 30 min at 4°C. The crosslinking reaction was stopped by the addition of 50 μL 1 M Tris-HCl, pH 7.5. Cells were washed in PBS and lysed in 100 μL M-PER (LifeTechnologies) for 15 min at 4°C. Cell debris and nuclei were removed by centrifugation and the post-nuclear supernatant was separated by gel electrophoresis on 3–8% Tris Acetate gels (LifeTechnologies). Proteins were transferred to nitrocellulose membrane and CD73 was visualized using anti-CD73 antibody EPR6115 (1:2,000 dilution; Abcam) and an HRP-conjugated goat-anti-rabbit IgG secondary antibody.
Protein-protein docking analysis
In silico modeling was performed using BioLuminate v1.9 (Shrödinger, LLC). A MEDI9447 Fv homology model was generated using PDB code 3h42 for the antibody variable region framework coordinates. 3h42 and MEDI9447 share 92% sequence identity in both the heavy and light chain framework with similarity scores of 1.0 and 0.96 for heavy and light framework, respectively; similarity is defined as the number of matching residues divided by the total number of residues; matching is defined as the 2 residues having a positive score according to the BLOSUM62 matrix.Citation57 CDR loop models were then generated using the BioLuminate loop database; the template loop structure chosen for each CDR was the largest cluster with the highest similarity with the MEDI9447 loop sequences. The identity and similarity of the CDR loops were as follows (identity, similarity): H1 (0.4, 0.8), H2 (0.82, 0.94), H3 (0.25, 0.38), L1 (0.85, 0.85), L2 (0.14, 0.43), L3 (0.18, 0.45). The top-ranked homology model (as designated by BioLuminate) was chosen for docking analysis. Protein docking was performed with the BioLuminate PIPER module using the CD73 structure coordinates from PDB code 4h2f and the MEDI9447 homology model. 70,000 antibody rotations were posed. All non-CDR residues (Chothia numbering) were excluded from the prediction; otherwise, no constraints or biases toward specific residues or regions of either the antibody or antigen were applied. The docking algorithm clusters spatially similar poses into a single pose structure; the top 30 clusters were evaluated for consistency with the epitope mapping mutagenesis studies and HDX results. Pose cluster 7 and 22 are shown in
. Model structures were generated using BioLuminate and PyMol.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_Datas.zip
Download Zip (3.2 MB)Acknowledgments
The authors would like to acknowledge Carl Hay and Qihui Huang (Department of Oncology Research, MedImmune) and Li Peng (ADPE, MedImmune) and Vaheh Oganesyan (ADPE, MedImmune) for helpful discussion and reagent sharing.
References
- Zimmermann H. 5′-nucleotidase: Molecular structure and functional aspects. Biochem J 1992; 285 (Pt 2):345–65; PMID:1637327; http://dx.doi.org/10.1042/bj2850345
- Sullivan JM, Alpers JB. In vitro regulation of rat heart 5′-nucleotidase by adenine nucleotides and magnesium. J Biol Chem 1971; 246:3057–63; PMID:4324346
- Kumar V. Adenosine as an endogenous immunoregulator in cancer pathogenesis: Where to go? Purinergic Signal 2013; 9:145–65; PMID:23271562; http://dx.doi.org/10.1007/s11302-012-9349-9
- Spychala J. Tumor-promoting functions of adenosine. Pharmacol Ther 2000; 87:161–73; PMID:11007998; http://dx.doi.org/10.1016/S0163-7258(00)00053-X
- Xu S, Shao QQ, Sun JT, Yang N, Xie Q, Wang DH, Huang QB, Huang B, Wang XY, Li XG, et al. Synergy between the ectoenzymes CD39 and CD73 contributes to adenosinergic immunosuppression in human malignant gliomas. Neuro Oncol 2013; 15:1160–72; PMID:23737488; http://dx.doi.org/10.1093/neuonc/not067
- Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007; 204:1257–65; PMID:17502665; http://dx.doi.org/10.1084/jem.20062512
- Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood 1997; 90:1600–10; PMID:9269779
- Eppell BA, Newell AM, Brown EJ. Adenosine receptors are expressed during differentiation of monocytes to macrophages in vitro. implications for regulation of phagocytosis. J Immunol 1989; 143:4141–5; PMID:2556476
- Leibovich SJ, Chen JF, Pinhal-Enfield G, Belem PC, Elson G, Rosania A, Ramanathan M, Montesinos C, Jacobson M, Schwarzschild MA, et al. Synergistic up-regulation of vascular endothelial growth factor expression in murine macrophages by adenosine A(2A) receptor agonists and endotoxin. Am J Pathol 2002; 160:2231–44; PMID:12057925; http://dx.doi.org/10.1016/S0002-9440(10)61170-4
- Ramanathan M, Pinhal-Enfield G, Hao I, Leibovich SJ. Synergistic up-regulation of vascular endothelial growth factor (VEGF) expression in macrophages by adenosine A2A receptor agonists and endotoxin involves transcriptional regulation via the hypoxia response element in the VEGF promoter. Mol Biol Cell 2007; 18:14–23; PMID:17065555; http://dx.doi.org/10.1091/mbc.E06-07-0596
- Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, Blackburn MR, Biaggioni I, Carbone DP, Feoktistov I, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 2008; 112:1822–31; PMID:18559975; http://dx.doi.org/10.1182/blood-2008-02-136325
- Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: A potent suppressor of antitumor immune responses. Trends Immunol 2012; 33:231–7; PMID:22487321; http://dx.doi.org/10.1016/j.it.2012.02.009
- Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res 1997; 57:2602–5; PMID:9205063
- Stagg J, Beavis PA, Divisekera U, Liu MC, Moller A, Darcy PK, Smyth MJ. CD73-deficient mice are resistant to carcinogenesis. Cancer Res 2012; 72:2190–6; PMID:22396496; http://dx.doi.org/10.1158/0008-5472.CAN-12-0420
- Zhi X, Chen S, Zhou P, Shao Z, Wang L, Ou Z, Yin L. RNA interference of ecto-5′-nucleotidase (CD73) inhibits human breast cancer cell growth and invasion. Clin Exp Metastasis 2007; 24:439–48; PMID:17587186; http://dx.doi.org/10.1007/s10585-007-9081-y
- Zhi X, Wang Y, Zhou X, Yu J, Jian R, Tang S, Yin L, Zhou P. RNAi-mediated CD73 suppression induces apoptosis and cell-cycle arrest in human breast cancer cells. Cancer Sci 2010; 101:2561–9; PMID:20874842; http://dx.doi.org/10.1111/j.1349-7006.2010.01733.x
- Bavaresco L, Bernardi A, Braganhol E, Cappellari AR, Rockenbach L, Farias PF, Wink MR, Delgado-Canedo A, Battastini AM. The role of ecto-5′-nucleotidase/CD73 in glioma cell line proliferation. Mol Cell Biochem 2008; 319:61–8; PMID:18636315; http://dx.doi.org/10.1007/s11010-008-9877-3
- Wang L, Zhou X, Zhou T, Ma D, Chen S, Zhi X, Yin L, Shao Z, Ou Z, Zhou P. Ecto-5′-nucleotidase promotes invasion, migration and adhesion of human breast cancer cells. J Cancer Res Clin Oncol 2008; 134:365–72; PMID:17671792; http://dx.doi.org/10.1007/s00432-007-0292-z
- Naito Y, Lowenstein JM. 5′-nucleotidase from rat heart. Biochemistry 1981; 20:5188–94; PMID:6271180; http://dx.doi.org/10.1021/bi00521a014
- Airas L, Niemela J, Salmi M, Puurunen T, Smith DJ, Jalkanen S. Differential regulation and function of CD73, a glycosyl-phosphatidylinositol-linked 70-kD adhesion molecule, on lymphocytes and endothelial cells. J Cell Biol 1997; 136:421–31; PMID:9015312; http://dx.doi.org/10.1083/jcb.136.2.421
- Knapp K, Zebisch M, Pippel J, El-Tayeb A, Muller CE, Strater N. Crystal structure of the human ecto-5′-nucleotidase (CD73): Insights into the regulation of purinergic signaling. Structure 2012; 20:2161–73; PMID:23142347; http://dx.doi.org/10.1016/j.str.2012.10.001
- Knofel T, Strater N. Mechanism of hydrolysis of phosphate esters by the dimetal center of 5′-nucleotidase based on crystal structures. J Mol Biol 2001; 309:239–54; PMID:11491293; http://dx.doi.org/10.1006/jmbi.2001.4656
- Schultz-Heienbrok R, Maier T, Strater N. A large hinge bending domain rotation is necessary for the catalytic function of escherichia coli 5′-nucleotidase. Biochemistry 2005; 44:2244–52; PMID:15709736; http://dx.doi.org/10.1021/bi047989c
- Knofel T, Strater N. E. coli 5′-nucleotidase undergoes a hinge-bending domain rotation resembling a ball-and-socket motion. J Mol Biol 2001; 309:255–66; PMID:11491294; http://dx.doi.org/10.1006/jmbi.2001.4657
- Ipata PL. Studies on the inhibition by nucleoside-triphosphates of sheep brain 5-nucleotidase. Biochem Biophys Res Commun 1967; 27:337–43; PMID:6035114; http://dx.doi.org/10.1016/S0006-291X(67)80103-7
- Burger RM, Lowenstein JM. Preparation and properties of 5′-nucleotidase from smooth muscle of small intestine. J Biol Chem 1970; 245:6274–80; PMID:4320834
- Iqbal J, Saeed A, Raza R, Matin A, Hameed A, Furtmann N, Lecka J, Sevigny J, Bajorath J. Identification of sulfonic acids as efficient ecto-5′-nucleotidase inhibitors. Eur J Med Chem 2013; 70:685–91; PMID:24215819; http://dx.doi.org/10.1016/j.ejmech.2013.10.053
- Raza R, Saeed A, Lecka J, Sevigny J, Iqbal J. Identification of small molecule sulfonic acids as ecto-5′-nucleotidase inhibitors. Med Chem 2012; 8:1133–9; PMID:22741787; http://dx.doi.org/10.2174/1573406411208061133
- Braganhol E, Tamajusuku AS, Bernardi A, Wink MR, Battastini AM. Ecto-5′-nucleotidase/CD73 inhibition by quercetin in the human U138MG glioma cell line. Biochim Biophys Acta 2007; 1770:1352–9; PMID:17643826; http://dx.doi.org/10.1016/j.bbagen.2007.06.003
- Gagliardi AR, Kassack M, Kreimeyer A, Muller G, Nickel P, Collins DC. Antiangiogenic and antiproliferative activity of suramin analogues. Cancer Chemother Pharmacol 1998; 41:117–24; PMID:9443624; http://dx.doi.org/10.1007/s002800050717
- Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, Dwyer KM, Smyth MJ. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A 2010; 107:1547–52; PMID:20080644; http://dx.doi.org/10.1073/pnas.0908801107
- Rust S, Guillard S, Sachsenmeier K, Hay C, Davidson M, Karlsson A, Karlsson R, Brand E, Lowne D, Elvin J, et al. Combining phenotypic and proteomic approaches to identify membrane targets in a ‘triple negative’ breast cancer cell type. Mol Cancer 2013; 12:11:4598-12-11; PMID:23406016; http;//dx.doi.org/10.1186/1476-4598-12-11
- Terp MG, Olesen KA, Arnspang EC, Lund RR, Lagerholm BC, Ditzel HJ, Leth-Larsen R. Anti-human CD73 monoclonal antibody inhibits metastasis formation in human breast cancer by inducing clustering and internalization of CD73 expressed on the surface of cancer cells. J Immunol 2013; 191:4165–73; PMID:24043904; http://dx.doi.org/10.4049/jimmunol.1301274
- Allard B, Turcotte M, Stagg J. Targeting CD73 and downstream adenosine receptor signaling in triple-negative breast cancer. Expert Opin Ther Targets 2014; 18:863–81; PMID:24798880; http://dx.doi.org/10.1517/14728222.2014.915315
- Sachsenmeier KF, Hay C, Brand E, Clarke L, Rosenthal K, Guillard S, Rust S, Minter R, Hollingsworth R. Development of a novel ectonucleotidase assay suitable for high-throughput screening. J Biomol Screen 2012; 17:993–8; PMID:22522649; http://dx.doi.org/10.1177/1087057112443987
- Hay C, Sult E, Huang Q, Hammond S, Mulgrew K, McGlinchey K, Fuhrmann S, Rothstein R, Poon E, Stewart R, Hollingsworth R, Sachsenmeier K. MEDI9447: enhancing anti-tumor immunity by targeting CD73 In the tumor microenvironment. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18–22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl): Abstract nr 285.
- Burger RM, Lowenstein JM. 5′-nucleotidase from smooth muscle of small intestine and from brain. inhibition of nucleotides. Biochemistry 1975; 14:2362–6; PMID:1169962; http://dx.doi.org/10.1021/bi00682a014
- Parker CH, Morgan CR, Rand KD, Engen JR, Jorgenson JW, Stafford DW. A conformational investigation of propeptide binding to the integral membrane protein gamma-glutamyl carboxylase using nanodisc hydrogen exchange mass spectrometry. Biochemistry 2014; 53:1511–20; PMID:24512177; http://dx.doi.org/10.1021/bi401536m
- Wei H, Mo J, Tao L, Russell RJ, Tymiak AA, Chen G, Iacob RE, Engen JR. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: Methodology and applications. Drug Discov Today 2014; 19:95–102; PMID:23928097; http://dx.doi.org/10.1016/j.drudis.2013.07.019
- Marcsisin SR, Narute PS, Emert-Sedlak LA, Kloczewiak M, Smithgall TE, Engen JR. On the solution conformation and dynamics of the HIV-1 viral infectivity factor. J Mol Biol 2011; 410:1008–22; PMID:21763503; http://dx.doi.org/10.1016/j.jmb.2011.04.053
- Oganesyan V, Peng L, Damschroder MM, Cheng L, Sadowska A, Tkaczyk C, Sellman BR, Wu H, Dall'Acqua WF. Mechanisms of neutralization of a human anti-alpha-toxin antibody. J Biol Chem 2014; 289:29874–80; PMID:25210036; http://dx.doi.org/10.1074/jbc.M114.601328
- Peng L, Oganesyan V, Wu H, Dall'Acqua WF, Damschroder MM. Molecular basis for antagonistic activity of anifrolumab, an anti-interferon-alpha receptor 1 antibody. MAbs 2015; 7:428–39; PMID:25606664; http://dx.doi.org/10.1080/19420862.2015.1007810
- Charrie A, Charriere G, Guerrier A. Hook effect in immunometric assays for prostate-specific antigen. Clin Chem 1995; 41:480–1; PMID:7533675
- Klemens MR, Sherman WR, Holmberg NJ, Ruedi JM, Low MG, Thompson LF. Characterization of soluble vs membrane-bound human placental 5′-nucleotidase. Biochem Biophys Res Commun 1990; 172:1371–7; PMID:2173922; http://dx.doi.org/10.1016/0006-291X(90)91601-N
- Piec G, Le Hir M. The soluble ‘low-km’ 5′-nucleotidase of rat kidney represents solubilized ecto-5′-nucleotidase. Biochem J 1991; 273(Pt 2):409–13; PMID:1846740; http://dx.doi.org/10.1042/bj2730409
- Brinkley BR, Beall PT, Wible LJ, Mace ML, Turner DS, Cailleau RM. Variations in cell form and cytoskeleton in human breast carcinoma cells in vitro. Cancer Res 1980; 40:3118–29; PMID:7000337
- Lamba V, Ghosh I. New directions in targeting protein kinases: Focusing upon true allosteric and bivalent inhibitors. Curr Pharm Des 2012; 18:2936–45; PMID:22571662; http://dx.doi.org/10.2174/138161212800672813
- Grover AK. Use of allosteric targets in the discovery of safer drugs. Med Princ Pract 2013; 22:418–26; PMID:23711993; http://dx.doi.org/10.1159/000350417
- Baqi Y, Weyler S, Iqbal J, Zimmermann H, Muller CE. Structure-activity relationships of anthraquinone derivatives derived from bromaminic acid as inhibitors of ectonucleoside triphosphate diphosphohydrolases (E-NTPDases). Purinergic Signal 2009; 5:91–106; PMID:18528783; http://dx.doi.org/10.1007/s11302-008-9103-5
- Evans WH, Gurd JW. Properties of a 5′-nucleotidase purified from mouse liver plasma membranes. Biochem J 1973; 133:189–99; PMID:4721620; http://dx.doi.org/10.1042/bj1330189
- Goding JW. Ecto-enzymes: Physiology meets pathology. J Leukoc Biol 2000; 67:285–311; PMID:10733089
- Salmi M, Jalkanen S. Cell-surface enzymes in control of leukocyte trafficking. Nat Rev Immunol 2005; 5:760–71; PMID:16200079; http://dx.doi.org/10.1038/nri1705
- Groves M, Lane S, Douthwaite J, Lowne D, Rees DG, Edwards B, Jackson RH. Affinity maturation of phage display antibody populations using ribosome display. J Immunol Methods 2006; 313:129–39; PMID:16730741; http://dx.doi.org/10.1016/j.jim.2006.04.002
- Xiao X, Chen Y, Mugabe S, Gao C, Tkaczyk C, Mazor Y, Pavlik P, Wu H, Dall'Acqua W, Chowdhury PS. A novel dual expression platform for high throughput functional screening of phage libraries in product like format. PLoS One 2015; 10:e0140691; PMID:26468955; http://dx.doi.org/10.1371/journal.pone.0140691
- Oganesyan V, Gao C, Shirinian L, Wu H, Dall'Acqua WF. Structural characterization of a human fc fragment engineered for lack of effector functions. Acta Crystallogr D Biol Crystallogr 2008; 64:700–4; PMID:18560159; http://dx.doi.org/10.1107/S0907444908007877
- Houde D, Berkowitz SA, Engen JR. The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J Pharm Sci 2011; 100:2071–86; PMID:21491437; http://dx.doi.org/10.1002/jps.22432
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A 1992; 89:10915–9; PMID:1438297; http://dx.doi.org/10.1073/pnas.89.22.10915