
ABSTRACT
The market for biotherapeutic monoclonal antibodies (mAbs) is large and is growing rapidly. However, attrition poses a significant challenge for the development of mAbs, and for biopharmaceuticals in general, with large associated costs in resource and animal use. Termination of candidate mAbs may occur due to poor translation from preclinical models to human safety. It is critical that the industry addresses this problem to maintain productivity. Though attrition poses a significant challenge for pharmaceuticals in general, there are specific challenges related to the development of antibody-based products. Due to species specificity, non-human primates (NHP) are frequently the only pharmacologically relevant species for nonclinical safety and toxicology testing for the majority of antibody-based products, and therefore, as more mAbs are developed, increased NHP use is anticipated. The integration of new and emerging in vitro and in silico technologies, e.g., cell- and tissue-based approaches, systems pharmacology and modeling, have the potential to improve the human safety prediction and the therapeutic mAb development process, while reducing and refining animal use simultaneously. In 2014, to engage in open discussion about the challenges and opportunities for the future of mAb development, a workshop was held with over 60 regulators and experts in drug development, mechanistic toxicology and emerging technologies to discuss this issue. The workshop used industry case-studies to discuss the value of the in vivo studies and identify opportunities for in vitro technologies in human safety assessment. From these and continuing discussions it is clear that there are opportunities to improve safety assessment in mAb development using non-animal technologies, potentially reducing future attrition, and there is a shared desire to reduce animal use through minimised study design and reduced numbers of studies.
Introduction
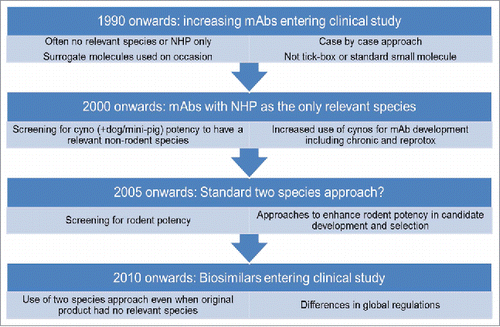
The market for protein-based biotherapeutics is large and is still growing rapidly. In 2015, 27% (12/45) of the drugs approved by the Food and Drug Administration (FDA) were biologic products, the highest number yet.Citation1 The largest group of biologics are antibody-based, mainly mAbs. There are 61 antibody-based products currently approved and in review in Europe and the US (as of October 2016).Citation2,Citation3 Recently, biosimilar products (a biologic medicinal product that contains a highly similar but not identical version of the original active substance of an already authorized original biologic medicinal product) have also entered the market. The European Medicines Agency (EMA) has approved 21 biosimilars to date,Citation2 including the first mAb biosimilar of infliximab, Inflectra (infliximab-dyyb), which received regulatory approval in Europe in September 2013.Citation4 In March 2015, the FDA approved its first biosimilar product (Zarxio™, filgrastim-sndz, Sandoz), which was followed by the first US mAb biosimilar approval, in April 2016 (for Inflectra).Citation5,Citation6 It is well documented that animal use in the development of mAbs, as well as other protein-based biotherapeutics and biosimilars, poses unique challenges to those associated with new chemical entities, and that these challenges have evolved over time and have contributed to attrition ().Citation7-11 While attrition owing to nonclinical safety events occurs less frequently for mAbs than for small molecules, these events certainly happen and underscore the importance of a thorough safety evaluation in relevant biologics systems.Citation12 Due to the species specificity of many products, non-human primates (NHPs) have been used for nonclinical safety and toxicology testing for the majority of antibody-based products as they are often the only species in which the mAb binds and has the desired pharmacological effect. However, there are often fundamental differences between primate and human physiology, and consequently there are often still deficiencies in the translation of NHP study results to human. For example, this may occur if the target does not play a role (or is redundant) in normal physiology in NHPs, or in cases where the target is still present, but has a different role or downstream effect in primate compared with human. In such situations, studies in NHP may therefore be of limited value to human risk assessment. Alternatively, mouse target knockout phenotypes can be used for hazard identification in place of the NHP, or surrogate molecules can be used in rodent species to demonstrate safety and efficacy.
Figure 1. Use of animals in mAb development and change in practice over time.
Toxicological science is also advancing rapidly across the public and private sectors. For example, in the chemicals industry adverse outcome pathways (AOPs), data from the USA's ToxCast program and exposure-based modeling are being applied to human risk assessment, shifting the focus toward human-specific mechanisms of action and pathways-based approaches.Citation13 There is regulatory interest in these approaches nationally and internationally at the Organization for Economic Co-operation and Development level (OECD).Citation14 Many of these approaches, in combination with technologies emerging from the research base, also show the potential to transform the pharmaceutical industry. For example, the ambitious collaboration between the National Institutes of Health's (NIH) National Center for Advancing Translational Science, Defense Advanced Research Projects Agency (DARPA) and FDA in the US has committed $150 million (from NIH and DARPA) over 5 y to develop tissue chips that mimic human physiology to screen for safe, effective drugs (http://www.ncats.nih.gov/tissuechip).Citation15 The FDA also recently announced a multi-year research and development agreement with Emulate Inc., a company founded by researchers at the Wyss Institute, to evaluate their “Organs-on-Chips” technology in laboratories at the agency's Center for Food Safety and Applied Nutrition.Citation16 The availability of new technologies alongside recent publications that have questioned whether the use of NHPs adds scientific value to the development of mAbs suggests that the timing is right to review the current biotherapeutic mAb development paradigm.Citation17,Citation18
In 2014, to engage in open discussion about the challenges and opportunities for the future of mAb development, a workshop was held with experts in drug development, mechanistic toxicology and emerging technologies such as cell and tissue-based approaches, systems pharmacology and modeling. The aims were to: 1) identify the knowledge and data gaps if scientists were to rely more heavily on the emerging technologies for the development of biotherapeutics; 2) determine how to optimise prediction of human safety by better understanding of mechanisms/target pharmacology; and 3) gain more value from fewer in vivo studies. The 60 participants included current FDA and European Union (EU) regulators and representatives from the pharmaceutical, biotechnology and contract research industries. A selection of current state-of-the-art techniques were showcased and discussed with a view to how these could be applied either now or in the future to improve the safety assessment of mAbs. Although, it was recognized it may be some time before any of these can be used successfully for decision-making in drug development, the workshop provided a unique opportunity to mine the vast knowledge and experience of this group to gain a consensus perspective on a future vision for safety assessment in mAb development. This paper provides an overview of the discussions that began at the workshop, descriptions of real-life industry case studies with consideration of the value of the in vivo and in vitro studies, and a plan for future work developed by the authors based on the output of the workshop and recent developments in the field.
The workshop
Emerging technologies
The most recent estimated figure for the cost of getting a drug to market is almost $2.6 billionCitation19 and between 2008 and 2010 productivity of the pharmaceutical industry was at an all-time low despite the introduction of the first wave of biotechnology derived products in the 1990s. There is some recent evidence that R&D productivity has turned a corner and the industry is sustainable again.Citation20,Citation21 However, attrition, which may be due to lack of efficacy as well as lack of translational safety, is still a huge problem, costing an estimated $1.4 billion per drug.Citation19 For mAbs, the lack of cross-reactivity in rodents may contribute to attrition as there are limited opportunities to study drug candidates in rodent pharmacology models. It is critical for the industry to reduce attrition in order for the increase in productivity to continue.
Many technologies are currently being used to reduce attrition in candidate screening for both efficacy and safety, including stem cells, cardiac assays, and in silico models. However, many of these are being used for small molecules, often to assess off-target toxicity, rather than for the screening of biotherapeutics.Citation22 One reason might be that such technologies are not relevant for mAb development because off-target toxicity rarely occurs and the toxicity is primarily related to specific on-target pharmacology. If this is true, the question remains as to whether these or similar technologies could be modified or developed to address questions that are more suited to biotherapeutics and, if so, how? Perhaps such technologies will need to be more case-specific, dependent on the binding target of the product.
Emerging technologies with possible relevance to biotherapeutic development were selected for discussion at the workshop and included: 1) ‘organs-on-chips’ types of in vitro technologies; 2) systems pharmacology and in silico modeling; 3) in vitro human immune models, as showcased by VaxDesigns's MIMIC® technology;Citation23 and 4) human pluripotent stem cells as a tool for developmental biology, as showcased by Stemina Biomarker Discovery's devTOX™ discovery assay.Citation24
The name ‘organ-on-a-chip’ refers to microfluidic cell culture devices that contain continuously perfused chambers inhabited by living cells arranged to simulate tissue- and organ-level physiology. The promise that ‘organs-on-chips’ offer to drug development has been well documented, but yet to be fully realized or adopted.Citation25,Citation26 This may be because these models are unable to fully represent the in vivo situation, to recreate a fully functioning organ outside of a living body, due to the more complex interplay between systems and processes at the whole animal level. Current research around the world is focused on the creation of reproducible systems that are representative of human disease states and also remain functional over a relevant time period to support target qualification and proof-of-concept studies. One of the most attractive components of the technology is the potential to generate genetically diverse ‘chips’ that may be used in clinical trial settings. The technology also enables high-resolution, real-time imaging and in vitro analysis of biochemical, genetic and metabolic activities of living human cells in a functional tissue and organ context.
Systems biology approaches aim to integrate the quantitative relationships between RNA, protein levels and metabolites to offer new insights into the function and behavior of organs, tissues and cells. Systems pharmacology describes an approach that links systems biology, pharmacology, medicinal chemistry and bioinformatics, and enables the development of models that predict and explain how drugs interact with biologic components. Modeling approaches are highly specific for the system(s) they describe and the questions being asked, thus, although they have shown value in defining potential on-target toxicities of new molecular entities, there is currently limited experience with using such approaches in the development of biologics.Citation27-29 Increased application of systems pharmacology and modeling for biologics could improve the characterization of the target (including target expression levels and expected pharmacokinetic/pharmacodynamic effects in in vivo studies), provide better data integration, and support the potential to reduce animal use. In part, the development of relevant in vitro assays with quantitative readouts in human primary cells and organs-on-chips will be instrumental in refining/applying these models for biologics. One successful example of an in vitro technique that has been applied to better characterize mAbs is the in vitro Comparative Immunogenicity Assessment (IVCIA).Citation30 This was developed as a tool for predicting potential relative immunogenicity of biotherapeutic mAbs as a screening and prioritization tool, to differentiate mAbs and detect differential immunogenicity as a result of aggregation, which has been shown to potentially enhance cytokine secretion and T-cell proliferation response in healthy volunteers.Citation31
Application of emerging technologies to real-life industry case studies
The assertion by van Meer et al. that NHPs do not necessarily add scientific value to the mAb development process is due mainly to the assumption that most mAb toxicity is related to exaggerated pharmacology and that such pharmacologically-mediated adverse effects could therefore be predicted from in vitro studies alone.Citation17,Citation18 However, since the time of the van Meer et al. publication, more exceptions to this assumption have been reported, in part as a result from data-sharing initiatives and workshops. One of the criticisms of publications reviewing current practice in how mAbs are developed is that they are often based solely on drugs that have been through regulatory review. Often, this approach is taken because regulatory dossiers (e.g., European public assessment reports, EU and FDA pharmacology and toxicology reviews) offer the only publically available information to assess. However, this leads to bias because conclusions are based on a limited sub-set of drugs, without representation of drugs that are terminated during development due to identified safety and toxicity issues. The drugs that are accepted for first-in-human (FIH) clinical studies are believed to be relatively safe drugs, as safety concerns such as severe toxicity would have been assessed non-clinically. To address this gap in available information, unpublished and published industry case-studies were gathered and analyzed to determine whether emerging technologies could have been used to predict nonclinical or clinical outcomes.
Several case-studies were selected for discussion at the workshop to enable a variety of targets and challenges to be debated in breakout groups. The same questions were asked for each case study and are listed in . Not all questions were relevant for all case studies and only relevant questions were answered in each breakout group.
Table 1. Questions for case studies addressed during the breakout sessions.
Workshop case studies
Case study 1: Anti-ADAMTS-5 mAb
Background
ADAMTS-5 is a member of the ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) protein family. It is an aggrecanase that degrades the aggrecan component of articular cartilage, making it an attractive target for osteoarthritis. Anti-ADAMTS-5 is a humanized, IgG1 Fc-disabled mAb that selectively inhibits ADAMTS-5 activity in the mouse, rat and NHP, including the cynomolgus monkey. The expression pattern of ADAMTS-5 shows that it is expressed in articular cartilage and surrounding joint tissues, but also many other tissues including arterial smooth muscle cells, mesothelium lining the peritoneal, pericardial and pleural cavities, smooth muscle cells in bronchi and pancreatic ducts, glomerular mesangial cells in the kidney, dorsal root ganglia, and Schwann cells.Citation32 ADAMTS-5 knockout mice show enlarged cardiac valves associated with accumulation of versican, which persists in adult mice.Citation33
Non-clinical development program and toxicology findings
Non-clinical toxicology studies were conducted in the cynomolgus monkey and also the Wistar Han rat (see ). Mechanistic investigational studies were performed in vitro (see ).
Table 2. In vivo studies for anti-ADAMTS-5.
Table 3. In vitro studies for anti-ADAMTS-5.
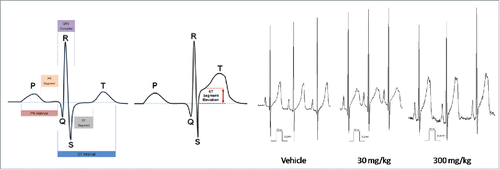
Cardiovascular effects (mean arterial pressure increase and ECG waveform abnormalities) were observed in monkey with anti-ADAMTS-5 in doses above 0.3 mg/kg. ST segment elevations were still detected 7 months post-dose (see ). Pre-existing knowledge of the target (i.e., phenotypes of knockout mice) suggested that there was a potential cardiovascular developmental risk, but did not predict the adverse electrophysiology. Furthermore, in vitro assays, such as the rabbit cardiac wedge assay and human CV ion channel assays did not detect the risk. There were no cardiovascular changes detected in the rat.
Figure 2. Dose-dependent ST segment elevation with anti-ADAMTS-5 in cynomolgus monkey.
Workshop breakout discussion
Attendees at the workshop were asked to consider the questions in . The consensus of the breakout group was that the cardiovascular effects observed in the nonclinical studies were not predictable based on the mechanism of action of anti-ADAMTS-5. Due to the target expression in the cardiovascular tissue, the heart was identified as a potential target organ; however, the observed effects (hemorrhage in the initial non-GLP study, acute arrhythmias, persistent ST segment elevations and dose-dependent delayed onset increase in blood pressure) could not have been predicted. The only adverse effect that could possibly be attributed to a background lesion in the animal was the hemorrhage.
Because a main focus of the workshop was the increased use of emerging technologies, the ability of in vitro approaches to identify the adverse effects was discussed. For anti-ADAMTS-5, the currently available in vitro tools were considered unable to predict the observed effects, as there are no in vitro models for hemodynamics or potential secondary pharmacologic effects, such as those on the extracellular matrix, that were relevant to this case study.
Evaluating the totality of the in vivo findings for this case study highlighted that the most useful information was obtained from the dedicated safety pharmacology evaluation in cynomolgus monkeys. Limitations of cardiovascular measurements conducted as part of the repeat-dose GLP toxicology study were also discussed and considered insufficient to detect the ECG findings observed in the safety pharmacology study. This was thought to be due to readings being taken at one minute intervals rather than over 24 hours as in the dedicated safety pharmacology study, and that there was no measurement of blood pressure included.
Case study 2: Anti-DLL4 mAb
Background
Delta-like ligand 4 (DLL4) is a ligand in the Notch family of endothelial cell receptors that functions to control the balance of tip and stalk cells during normal vascular development. Heterozygous DLL4 knockout mice show embryonic lethality due to vascular abnormalities, and more recent experiments show that conditional knockout of Notch 1 can also lead to the development of vascular tumors in mice.Citation34,Citation35 Targeting DLL4 with an antibody (anti-DLL4 IgG1) yielded robust anti-tumor activity in several nonclinical models, making DLL4 an attractive therapeutic target.Citation36-39 The findings from in vivo toxicology studies using an anti-DLL4 IgG1 mAb, however, raised serious safety concerns that were considered target-related based on the known expression and function of DLL4, although the specific manifestations of toxicity observed following repeated treatment had not been predicted a priori from pre-existing information alone. Given the potential promise of DLL4 inhibition for anti-tumor activity, a second approach was then undertaken by engineering a Fab’2 fragment to target DLL4 (rather than inhibition of DLL4 with the full IgG1 antibody). Although the Fab’2 approach ultimately mitigated some of the target-related toxicities seen with the IgG1 molecule, other unexpected target-related toxicities were revealed and resulted in termination of the program before FIH studies.Citation40
Non-clinical development program and toxicology findings
Various in vitro studies were performed with anti-DLL4 IgG1 and Fab’2 candidate molecules to confirm effective DLL4 pathway inhibition, including a mouse retinal explant model and a human umbilical vein endothelial cell potency assay to demonstrate the expected pharmacology of DLL4 inhibition of nonproductive angiogenesis and endothelial cell proliferation, respectively. Biacore data confirmed similar potency of anti-DLL4 binding across mice, rats, NHP and humans. GLP toxicology studies were therefore conducted using the Sprague Dawley rat and the cynomolgus monkey for both the IgG1 mAb and the Fab’2 candidate molecules, to evaluate toxicity in 2 species (rodent and non-rodent) as per ICH S6.Citation41 Findings observed following anti-DLL4 IgG1 mAb administration in both species included marked atrophy of centrilobular hepatic cords, sinusoidal dilation, bile ductular proliferation, elevated liver function tests and decreased red blood cells. However, the severity and incidence of these findings differed between species, such that the liver findings were more severe in the rat relative to the cynomolgus monkey, and the decrease in red blood cells was more severe in the cynomolgus monkey. There were also additional findings in the rat that were not observed in NHPs, including proliferative vascular neoplasms in the skin, lung, and heart ().
Table 4. In vivo studies for anti-DLL4 IgG1 mAb.
To further evaluate whether the DLL4 pathway could be therapeutically targeted without compromising safety, a Fab’2 fragment of the anti-DLL4 antibody with more rapid clearance and shorter half-life than the original IgG1 antibody was developed.Citation34 GLP toxicology studies with the new molecule showed an improved toxicity profile regarding the liver, red blood cell loss, and occurrence of proliferative vascular lesions. However, new findings were also identified with the Fab’2 fragment in the heart and lung of both species that were suggestive of pulmonary hypertension and considered related to DLL4 inhibition (). Together, these findings suggest that inhibiting the DLL4 pathway under different conditions (e.g., different exposure regimens) may lead to differential and unexpected findings in vivo.
Table 5. In vivo studies for anti-DLL4 Fab’2 fragment.
Workshop breakout discussion
At the start of the anti-DLL4 development program, the hypothesis was that the DLL4 pathway was only active in tumor, rather than normal vasculature, and that the effects would be similar to the approved angiogenesis inhibitor, bevacizumab (Avastin®). Attendees were asked to consider the questions in . The breakout group considered that clinically relevant findings in general repeat dose toxicity studies were related to the mechanism of action of the mAb and the majority of the participants agreed with this. However, it was agreed that only some of the findings, for example those that were characteristic of bevacizumab, were predictable a priori. There were significant, clinically relevant toxicities (e.g., liver sinusoidal changes, heart and lung changes and anemia) that the participants thought were not predictable before in vivo studies. Importantly, the dose-related toxicities seen in rats and monkeys resulted in a decision to terminate both anti-DLL4 molecules before FIH studies.
Only a third (30%) of the participants thought that some of these clinically relevant findings could be predictable in the future using existing/future in vitro technologies and systems biology approaches. Important gaps identified in currently available in vitro approaches included difficulty in modeling paracrine effects between at least 2 inter-regulated cell types, as well as in modeling potential hemodynamic effects that could lead to pathway-related changes only apparent in an in vivo setting. For example, data from human cardiomyocytes or hepatocytes would be limited as anti-DLL4 may be acting at the endothelium and the sinusoids, respectively. A primary limitation of current in vitro systems is therefore associated with the ability to integrate multiple cell types (e.g., stalk cells, tip cells and epithelia), with appropriate tissue architecture and hemodynamics, to tackle questions relating to potential in vivo physiologic effects of the drug. The potential for Notch signaling studies to provide information on cross-species potency and add value to the overall program was also discussed. Although signaling studies were considered to be of limited value in predicting in vivo toxicity in this case, the group were interested in evaluating the potential for 3-dimensional (3D) tissue models, including novel microfluidic and dynamic flow systems currently in development (e.g., as described in ref. Citation42), to predict toxicity within a more physiologic organ architecture. Given the liver phenotype observed following administration of anti-DLL4 in vivo, these more complex organ models were thought to hold potentially greater promise for accurate prediction of toxicities that may only be reproduced in the context of relevant sinusoidal architecture or hemodynamic changes on vascular endothelial cells. The breakout group agreed that if development of an anti-DLL4 molecule had continued further, a chronic toxicity study in the rat alone (rather than the cynomolgus monkey or in both species) may be useful to further identify potential effects with long-term treatment, as the rat and monkey exhibited similar toxicity profiles, and it would therefore be appropriate to conduct additional nonclinical studies in the lower-order species to limit NHP use.Citation41
Case study 3: Anti-amyloid β mAb
Background
The anti-amyloid β mAb discussed in case study 3 is a humanized monoclonal IgG1 antibody. It is specific for a conformation of amyloid β (Aβ) protein oligomer and binds the oligomer with high selectivity compared with other Aβ conformations such as fibrils or monomer. This high selectivity for the Aβ oligomer was predicted to result in improved efficacy and reduced side effects in the treatment of Alzheimer's Disease. The target antigen is primarily present in Alzheimer's Disease state, and is essentially undetectable in normal animals. Unexpected cross-reactivity of the anti-amyloid β mAb to a plasma protein cytokine resulted in preclinical toxicity in NHPs.Citation43 Studies by Vugmeyster et al., which used a humanized anti-amyloid antibody against amino acids 3-6 of primate amyloid beta, and which was published after this work had been performed, demonstrated off-target binding to fibrinogen which was shown to slow clearance.Citation44
Non-clinical development program and toxicology findings
The in vivo and in vitro studies are summarized in and , respectively. The anti-amyloid β mAb was found to normalize synaptic function and improve cognitive function in amyloid precursor protein (APP) transgenic mouse model of Alzheimer's Disease. No side effects were observed in a 4-week non-clinical APP mouse study. A tissue cross-reactivity panel in monkeys and humans showed no noticeable binding, and there was no binding to human peripheral blood cells. No side effects were observed following a single low-dose administration in a cynomolgus monkey pharmacokinetics study, but severe toxicological effects were observed in a 13-week repeat-dose cynomolgus monkey study. At low doses (20 and 60 mg/kg/week), thrombocytopenia and vasculature changes (medial hypertrophy and thrombosis) were observed, along with neuron loss and microhemorrhages in the brain. Higher doses (120 and 200 mg/kg) caused an acute infusion reaction upon the first dose, with lethal consequences. The rapid onset after the first dose at 200 mg/kg was indicative of an effect initiated by binding of anti-amyloid β mAb to an already present plasma antigen. Further evaluations identified unintended off-target binding to a specific plasma protein cytokine that is released from activated platelets and has strong chemoattractant properties for neutrophils and fibroblasts (). The binding of the anti-amyloid β mAb to the plasma protein resembles the pathological function of Heparin-induced Thrombocytopenia (HIT), leading to HIT-like symptoms such as thrombocytopenia. Thus, the off-target binding was consistent with the thrombocytopenia, vascular changes and infusion reactions that were observed in the cynomolgus monkey toxicity study.
Table 6. In vivo studies for anti-amyloid β mAb.
Table 7. In vitro studies for anti-amyloid β mAb.
Workshop breakout discussion
The consensus of the breakout group was that the effects observed in the nonclinical studies, particularly the thrombocytopenia and vascular effects, were not predictable based on the mechanism of action of anti-amyloid β mAb. The off-target binding could not have been identified via any other means of in vitro testing available at the time (e.g., cytokine release, whole blood binding assays), though it may now be possible to identify potential off-target binding through an extended in vitro binding cascade. A wider range of in vitro approaches may have aided the prediction of the effects observed, for example to screen for plasma or serum component binding before in vivo studies.
Over half of the participants (53%) agreed that in future the observed effects could be predictable with greater use of new/existing in vitro technologies, whereas a third of the participants (30%) disagreed, with the remaining participants undecided. Since the off-target cross-reactivity was only present in human and cynomolgus plasma, not in mouse, rat or dog plasma, this case demonstrates the importance of testing the safety of therapeutic antibodies in a species relevant for both on-target and off-target binding.
Workshop consensus
A voting system was used throughout the workshop to gauge participant opinion, and the results are presented and discussed below.
Opportunities for in vitro technologies
The presentations and discussion at the workshop inspired many participants to think differently about how new technologies could be integrated into drug development approaches for biotherapeutics, and most (83%) thought that industry should use more in vitro approaches wherever feasible to reduce animal use. Almost all participants (95%) agreed that there were specific situations where in vitro data from human systems was more important than in vivo data from animal studies (e.g., cytokine release (TGN1412)). When asked whether regulators would accept in vitro data in lieu of some in vivo data to support FIH dosing, 55% of participants were sceptical, pointing toward the need for the scientific community to generate convincing data on the validity of in vitro models for human risk assessment. The most likely aspect of nonclinical toxicology for biopharmaceuticals to be replaced by in vitro approaches was thought to be carcinogenicity (50%), followed by general toxicology (29%) then reproductive toxicology (11%) and juvenile toxicology (11%). The need to improve screening for off-target tissue binding before in vivo studies (and to potentially replace GLP tissue cross-reactivity studies in the future) was also identified as an area ripe for improvement (83% agreement).
Value of the in vivo studies
The majority of participants (86%) agreed that while some in vivo nonclinical findings resulting in termination of projects may have been false positives and not relevant to humans, these were difficult to predict and it was unlikely that such risk would readily be taken to enable these drugs to enter the clinic. Many companies also reported increased requests for juvenile toxicity studies to support pediatric clinical development. Participants generally agreed that these studies were rarely or never needed to support pediatric indications for age 6-12 y (never (61%), sometimes (34%) and always (5%)). There was a slight shift in participant experience for the 2-6 y age range, with half workshop participants voting that these studies were rarely or never needed (never (49%), sometimes (46%) and always (5%)). Better integration of information from general toxicology studies, clinical data from adult patients, modeling and systems biology approaches (such as those currently used for dose calculation) was considered likely to supersede the need for juvenile toxicology studies (73% agreed). This position is supported in the newly developed ICH S11 ‘Non-clinical Safety Testing in Support of Development of Pediatric Medicines’ concept paper that provides guidance and direction on the nonclinical safety studies needed to support a pediatric development program.Citation45 Companies had also experienced regulatory requests to assess bone quality endpoints in ovariectomized NHPs for certain classes of drugs, although participants generally agreed that this type of study did not add value for human risk assessment (54% agreed, 17% disagreed, 29% don't know).
One approach to refine in vivo nonclinical development programmes in the future may be to conduct a single toxicology study to enable clinical trials. This study would not need to be longer than 6 months as long-term chronic toxicology studies (9/12 months) do not often detect additional/new toxicities compared with the shorter-term studies, as agreed upon with the ICH S6 Addendum.Citation11,Citation41,Citation46 More data are required to assess whether there are more scientifically justified opportunities to conduct a single study of 12 weeks for FIH clinical trials other than those for serious life-threatening conditions. Indeed, in cases where the mAb is directed against a target that is minimally expressed in naïve animals or does not play a role in normally physiology, a short-term study of 1 month duration may be sufficient. The current ICH guidelines and regulatory environment should be amenable to this, as the guidelines are meant to act as a guide, and do not currently dictate study duration, aside from that they should be based on the intended duration of clinical exposure and disease indication. Furthermore, regulators will allow deviation from the guidelines, taking scientific rationale into account on a case-by-case basis.
Data-sharing and transparency
There was consensus from regulators and industry that the workshop had provided a useful forum for open discussion of case studies that were not in the public domain, and participants agreed that there was value in increasing availability of data from terminated biotherapeutics to regulators (91% agreed). There would also be value in making this information available to other industry stakeholders to reduce redundancy in animal studies and potentially enable broader innovation across the industry. However, the significant challenge identified would be achieving this in practice, as only 55% of attendees had confidence that they could persuade their companies to see the value of releasing such cases into the public domain due to competitive and intellectual property concerns. Furthermore, there are often difficulties in publishing this sort of data if the project has been terminated before establishing the exact cause of the toxicity.
The ongoing challenge for regulators is that they only see the few molecules that companies choose to advance into clinical trials, which are typically much less likely to have associated or severe toxicities, as the most concerning candidate molecules/targets have often been terminated before any regulatory interactions. In developing a future vision for mAb development, one important aspect for consideration is a continued evolution of regulatory practice and policy. For example, the amount of knowledge and data that is generated as a by-product of the regulatory submissions process is critical to ensure future strategy is directed and informed by science through a broad evidence-base. Although information collected in surveys is useful for certain purposes, such as in developing recommendations on good practices, in this case the importance of detailed specific case study information was acknowledged.
Discussion and future work
The scientific and regulatory community clearly share a vision for continued evaluation and integration of emerging technologies to reduce and refine animal use for biotherapeutic mAb development. However, there are still several barriers that must be recognized and overcome to make this a reality. When individual case studies were discussed and retrospectively analyzed, the ability of existing in vitro and in silico technologies to detect or predict toxicities observed in in vivo studies was noted as lacking (summary in ). Some of the observed in vivo effects, such as changes in blood pressure or paracrine effects, would not have been predicted using currently available technologies; therefore, the challenge for the future will be to advance and apply novel technologies that have the capability to more closely represent the in vivo situation.
Table 8. Summary of case-study data and the ability of existing in vitro and in silico technologies to detect or predict toxicities observed in the in vivo studies.
The majority of clinically relevant findings for mAbs are based on their mechanism of action. However, the toxicities presented in the case studies were, in general, not predictable before in vivo studies despite their relationship to the pharmacological action of the mAb. It is also important to note that many associated clinical toxicities such as some cancers, progressive multifocal leukoencephalopathy and infection, are so rare that they are not realistically detected in any in vitro or in vivo study. Currently, the field remains insufficiently confident in the ability of in vitro models to capture unpredictable toxicological findings as highlighted in the case studies, although there is much enthusiasm, commitment, and perceived potential for the industry to work towards this aim. Significant activity will be required to progress this field to be able to confidently predict unexpected toxicities from in vitro models. The development of more sophisticated and relevant in vitro technologies for safety assessment of mAbs may need to be more case-dependent, to take in to account their innate complexity, diversity and size, as well as their specific mechanism of action. A major recommendation of the participants at the workshop was for the establishment of a framework that could improve pre-competitive data-sharing between companies developing biologic products. Increased communication and data-sharing would enhance progress, increase understanding between industry and regulators, and support advancement toward common goals. The challenge faced in developing such a framework is in providing incentives for companies to share data on terminated compounds, which could take the form of individual company publications, cross-company initiatives, consideration of coordination with the EMA safe harbour effort, as well as development of an online journal, database or repository that would provide an easily accessible platform to share additional case studies. The NC3Rs could potentially serve as an honest broker to take this type of initiative forward; Biosafe (a committee within the Biotechnology Innovation Organization, a trade association for biotechnology-related organizations globally) is also working to collect several similar case studies that can be published and presented to the FDA.
As well as consideration of the potential for emerging technologies, the value of the existing in vivo studies was also discussed at the workshop. A topic identified with the potential to unnecessarily increase NHP use in the future was an increase in juvenile toxicity studies as default practice to support mAb development in pediatric populations, due to regulatory perception within companies and previous requests from the Pediatric Committee (PDCO/EMA). The regulatory requests were not always deemed to be scientifically driven and many participants disagreed that juvenile toxicity studies were necessary to inform pediatric safety in many cases, as there is potential to better utilize and integrate information from general toxicology studies and clinical data from adult patients. Since the discussions at the workshop, the guidance published in the ICH S11 concept paper may alleviate some of these concerns.Citation45 However, to prevent unnecessary conduct of these studies as the general rule, a data-sharing initiative will be needed to evaluate whether juvenile toxicity studies in animals provide any additional clinically relevant information, and if so, in what circumstances.
A future vision for mAb development is one in which fewer animals are used, but where the data obtained are more predictive of human safety. Therefore the typical approach to safety assessment of mAbs was considered. Typically two studies, one to support FIH clinical studies (IND-enabling) and one to support registration, are performed during mAb development. In some cases for oncology indications, a single study may suffice.Citation47 However, an alternate approach could be to use a single, comprehensive in vivo study for the majority of mAbs that includes, in addition to toxicity endpoints, relevant pharmacodynamic, biomarker and potentially safety pharmacology endpoints. It has been argued that new clinically relevant findings are rarely identified in long-term studies that were not observed or could not have been predicted from the short-term study.Citation11,Citation46 Future work will involve whether both the 12-week and 26-week studies are of value in detecting clinically relevant findings. Of course, it is often the rare cases that ultimately drive regulations to ensure adequate safety in clinical trials. One recommendation for further progress in this area is to generate an evidence-base to help determine the frequency and types of toxicity that are observed only in long-term studies (6 months) compared with the shorter-term studies (1-3 months), and to explore whether these risks might be predicted in advance for specific types of targets. For example, the majority of participants felt that products such as cytokines and other soluble factors could be safely approved based on the IND-enabling toxicology study with no chronic toxicology (54% agreed, 32% disagreed, 15% don't know).
Conclusions
A number of areas have been identified for future resource and investment that are critical to reach a scientifically driven vision for future biotherapeutic mAb development. These include the development and increased use of emerging technologies such as cell and tissue-based technologies that are suitable for mAb development, and opportunities to waive chronic and juvenile toxicity studies. Currently, many of the emerging technologies are being developed with small molecule new molecular entities in mind, rather than biologics, and a shift toward application for biologics is needed for these technologies to play a role in mAb development (e.g., development of hemodynamic models or paracrine effects models is lacking). A number of organizations have an interest in progressing this area across the regulatory, industry and public sectors and the time is right for collaboration to shape future investment, data-sharing activities and technology development in this area. The aim of our workshop was to contemplate what will be possible in the next 10 y rather than focus on our current capabilities. It is clear that there are opportunities to improve mAb development, but this will not happen without the collective knowledge, experience and dedication of experienced drug safety professionals and a more open-minded approach to mAb development.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Mullard A. 2015 FDA drug approvals. Nat Rev Drug Discov 2016; 15(2):73–6; PMID:26837582; https://doi.org/10.1038/nrd.2016.15
- EMA. European public assessment reports, human medicines [Internet]. The European Medicines Agency [cited 2016 October 19]. 2016 Available from: http://www.ema.europa.eu/ema/index.jsp?curl = pages%2Fmedicines%2Flanding%2Fepar_search.jsp&mid=&searchTab = searchByAuthType&alreadyLoaded = true&isNewQuery = true&status = Authorised&keyword = Enter+keywords&searchType = name&taxonomyPath=&treeNumber=&searchGenericType = biosimilars&genericsKeywordSearch=Submit.
- FDA. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm 2016.
- EMA. http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2013/06/WC500144941.pdf. 2013.
- FDA. U.S. Food and Drug Administration Press Release. 6 March 2015: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm436648.htm. 2015.
- FDA. U.S. Food and Drug Administration Press Release. 5 April 2016: http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm494227.htm. 2016.
- Buckley LA, Chapman K, Burns-Naas LA, Todd MD, Martin PL, Lansita JA. Considerations regarding nonhuman primate use in safety assessment of biopharmaceuticals. Int J Toxicol 2011; 30(5):583-90; PMID:22013138; https://doi.org/10.1177/1091581811415875
- Bussiere JL. Species selection considerations for preclinical toxicology studies for biotherapeutics. Expert Opin Drug Metab Toxicol 2008; 4(7):871-7; PMID:18624676; https://doi.org/10.1517/17425255.4.7.871
- Chapman K, Pullen N, Coney L, Dempster M, Andrews L, Bajramovic J, Baldrick P, Buckley L, Jacobs A, Hale G, et al. Preclinical development of monoclonal antibodies: Considerations for the use of non-human primates. MAbs 2009; 1(5):505-16; PMID:20065651; https://doi.org/10.4161/mabs.1.5.9676
- Chapman K, Pullen N, Graham M, Ragan I. Preclinical safety testing of monoclonal antibodies: The significance of species relevance. Nat Rev Drug Discov 2007; 6(2):120-6; PMID:17268483; https://doi.org/10.1038/nrd2242
- Chapman KL, Andrews L, Bajramovic JJ, Baldrick P, Black LE, Bowman CJ, Buckley LA, Coney LA, Couch J, Maggie Dempster A, et al. The design of chronic toxicology studies of monoclonal antibodies: Implications for the reduction in use of non-human primates. Regul Toxicol Pharmacol 2012; 62(2):347-54; PMID:22100994; https://doi.org/10.1016/j.yrtph.2011.10.016
- Brennan FR, Cavagnaro J, McKeever K, Schutten M, Vahle J, Weinbauer G, Black L. Safety testing of monoclonal antibodies in non-human primates: Case studies highlighting their impact on risk assessment for humans. Ready for submission. 2017.
- Judson R, Kavlock R, Martin M, Reif D, Houck K, Knudsen T, Richard A, Tice RR, Whelan M, Xia M, et al. Perspectives on validation of high-throughput assays supporting 21st century toxicity testing. ALTEX 2013; 30(1):51-6; PMID:23338806; https://doi.org/10.14573/altex.2013.1.051
- https://aopwiki.org.
- NIH. http://www.nih.gov/news-events/news-releases/nih-darpa-fda-collaborate-develop-cutting-edge-technologies-predict-drug-safety. 2011.
- Emulate. https://emulatebio.com/press/fda-collab-agreement-emulate/. 2017.
- van Meer PJ, Kooijman M, Brinks V, Gispen-de Wied CC, Silva-Lima B, Moors EH, Schellekens H. Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. MAbs 2013; 5(5):810-6; PMID:23924803; https://doi.org/10.4161/mabs.25234
- van Meer PJ, Kooijman M, van der Laan JW, Moors EH, Schellekens H. The value of non-human primates in the development of monoclonal antibodies. Nat Biotechnol 2013; 31(10):882-3; PMID:24104750; https://doi.org/10.1038/nbt.2709
- DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: New estimates of R&D costs. J Health Econ 2016; 47:20-33; PMID:26928437; https://doi.org/10.1016/j.jhealeco.2016.01.012
- Scannell JW, Blanckley A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov 2012; 11(3):191-200; PMID:22378269; https://doi.org/10.1038/nrd3681
- Schulze U, Baedeker M, Chen YT, Greber D. R&D productivity: On the comeback trail. Nat Rev Drug Discov 2014; 13(5):331-2; PMID:24751818; https://doi.org/10.1038/nrd4320
- Kizhedath A, Wilkinson S, Glassey J. Glassey, applicability of predictive toxicology methods for monoclonal antibody therapeutics: Status Quo and scope. Arch Toxicol 2017; 91(4):1595-612; PMID:27766364; https://doi.org/10.1007/s00204-016-1876-7
- http://www.vaxdesign.com/mimic-technology.
- http://www.stemina.com.
- Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol 2014; 32(8):760-72; PMID:25093883; https://doi.org/10.1038/nbt.2989
- Borenstein JT. Organs-on-Chips: How microsystems technology can transform the drug development process. IEEE Pulse 2016; 7(2):22-6; PMID:26978847; https://doi.org/10.1109/MPUL.2015.2513722
- Jones HM, Chen Y, Gibson C, Heimbach T, Parrott N, Peters SA, Snoeys J, Upreti VV, Zheng M, Hall SD. Physiologically based pharmacokinetic modeling in drug discovery and development: A pharmaceutical industry perspective. Clin Pharmacol Ther 2015; 97(3):247-62; PMID:25670209; https://doi.org/10.1002/cpt.37
- Pelkonen O, Turpeinen M, Raunio H. in vivo-in vitro-in silico pharmacokinetic modelling in drug development: Current status and future directions. Clin Pharmacokinet 2011; 50(8):483-91; PMID:21740072; https://doi.org/10.2165/11592400-000000000-00000
- Valerio LG Jr. In silico toxicology for the pharmaceutical sciences. Toxicol Appl Pharmacol 2009; 241(3):356-70; PMID:19716836; https://doi.org/10.1016/j.taap.2009.08.022
- Joubert MK, Deshpande M, Yang J, Reynolds H, Bryson C, Fogg M, Baker MP, Herskovitz J, Goletz TJ, Zhou L, et al. Use of in vitro assays to assess immunogenicity risk of antibody-based biotherapeutics. PLoS One 2016; 11(8):e0159328; PMID:27494246; https://doi.org/10.1371/journal.pone.0159328
- Joubert MK, Hokom M, Eakin C, Zhou L, Deshpande M, Baker M, Goletz T, Kerwin B, Chirmule N, Narhi L, et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J Biol Chem 2012 Jul 20; 287(30):25266-79; PMID:22584577; https://doi.org/10.1074/jbc.M111.330902
- McCulloch DR, Le Goff C, Bhatt S, Dixon LJ, Sandy JD, Apte SS. Adamts5, the gene encoding a proteoglycan-degrading metalloprotease, is expressed by specific cell lineages during mouse embryonic development and in adult tissues. Gene Expr Patterns 2009; 9(5):314-23; PMID:19250981; https://doi.org/10.1016/j.gep.2009.02.006
- Dupuis LE, McCulloch DR, McGarity JD, Bahan A, Wessels A, Weber D, Diminich AM, Nelson CM, Apte SS, Kern CB. Altered versican cleavage in ADAMTS5 deficient mice; a novel etiology of myxomatous valve disease. Dev Biol 2011; 357(1):152-64; PMID:21749862; https://doi.org/10.1016/j.ydbio.2011.06.041
- Phng LK, Gerhardt H. Angiogenesis: A team effort coordinated by notch. Dev Cell 2009; 16(2):196-208; PMID:19217422; https://doi.org/10.1016/j.devcel.2009.01.015
- Liu Z, Turkoz A, Jackson EN, Corbo JC, Engelbach JA, Garbow JR, Piwnica-Worms DR, Kopan R. Notch1 loss of heterozygosity causes vascular tumors and lethal hemorrhage in mice. J Clin Invest 2011; 121(2):800-8; PMID:21266774; https://doi.org/10.1172/JCI43114
- Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, Fitch-Bruhns M, Lazetic S, Park IK, Sato A, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell 2009; 5(2):168-77; PMID:19664991; https://doi.org/10.1016/j.stem.2009.05.019
- Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006; 444(7122):1032-7; PMID:17183313; https://doi.org/10.1038/nature05355
- Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, Kowalski J, Watts RJ, Callahan C, Kasman I, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006; 444(7122):1083-7; PMID:17183323; https://doi.org/10.1038/nature05313
- Scehnet JS, Jiang W, Kumar SR, Krasnoperov V, Trindade A, Benedito R, Djokovic D, Borges C, Ley EJ, Duarte A, et al. Inhibition of Dll4-mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood 2007; 109(11):4753-60; PMID:17311993; https://doi.org/10.1182/blood-2006-12-063933
- Couch JA, Zhang G, Beyer JC, de Zafra CL, Gupta P, Kamath AV, Lewin-Koh N, Tarrant J, Allamneni KP, Cain G, et al. Balancing efficacy and safety of an anti-DLL4 antibody through pharmacokinetic modulation. Clin Cancer Res 2016; 22(6):1469-79; PMID:26589434; https://doi.org/10.1158/1078-0432.CCR-15-1380
- ICH. Preclincal safety evaluation of biotechnology-derived pharmaceuticals. International Conference on Harmonisation (ICH). Topic S6(R1). June 2011; S6(R1).
- Sistare FD, Mattes WB, LeCluyse EL. The promise of new technologies to reduce, refine, or replace animal use while reducing risks of drug induced liver injury in pharmaceutical development. ILAR J6 2016; 57(2):186-211; PMID:28053072; https://doi.org/10.1093/ilar/ilw025
- Barghorn S. Off target toxicity of an anti-amyloid beta antibody for Alzheimer's disease immunotherapy. Presented at IBC's Antibody Engineering & Therapeutics conference December 2013.
- Vugmeyster Y, Szklut P, Wensel D, Ross J, Xu X, Awwad M, Gill D, Tchistiakov L, Warner G. Complex pharmacokinetics of a humanized antibody against human amyloid beta peptide, anti-abeta Ab2, in nonclinical species. Pharm Res 2011; 28:1696-706. 45; PMID:21424161; https://doi.org/10.1007/s11095-011-0405-x
- ICH. Final concept paper S11: Nonclinical safety testing in support of development of pediatric medicines. International Council on Harmonisation (ICH). Topic S11. September 2014. Endorsed by the ICH Steering Committee on 10 November 2014., S11.
- Clarke J, Hurst C, Martin P, Vahle J, Ponce R, Mounho B, Heidel S, Andrews L, Reynolds T, Cavagnaro J. Duration of chronic toxicity studies for biotechnology-derived pharmaceuticals: Is 6 months still appropriate? Regul Toxicol Pharmacol 2008; 50(1):2-22; PMID:17998153; https://doi.org/10.1016/j.yrtph.2007.08.001
- ICH. Nonclinical evaluation for anticancer pharmaceuticals. International Conference on Harmonisation (ICH). Topic S9. March 2010; S9.