
ABSTRACT
A large body of data exists demonstrating that neonatal Fc receptor (FcRn) binding of an IgG via its Fc CH2-CH3 interface trends with the pharmacokinetics (PK) of IgG. We have observed that PK of IgG molecules vary widely, even when they share identical Fc domains. This led us to hypothesize that domains distal from the Fc could contribute to FcRn binding and affect PK. In this study, we explored the role of these IgG domains in altering the affinity between IgG and FcRn. Using a surface plasmon resonance-based assay developed to examine the steady-state binding affinity (KD) of IgG molecules to FcRn, we dissected the contributions of IgG domains in modulating the affinity between FcRn and IgG. Through analysis of a broad collection of therapeutic antibodies containing more than 50 unique IgG molecules, we demonstrated that variable domains, and in particular complementarity-determining regions (CDRs), significantly alter binding affinity to FcRn in vitro. Furthermore, a panel of IgG molecules differing only by 1–5 mutations in CDRs altered binding affinity to FcRn in vitro, by up to 79-fold, and the affinity values correlated with calculated isoelectric point values of both variable domains and CDR-L3. In addition, tighter affinity values trend with faster in vivo clearance of a set of IgG molecules differing only by 1–3 mutations in human FcRn transgenic mice. Understanding the role of CDRs in modulation of IgG affinity to FcRn in vitro and their effect on PK of IgG may have far-reaching implications in the optimization of IgG therapeutics.
Abbreviations
| ADA | = | anti-drug antibody |
| β2m | = | beta-2 microglobulin |
| CDRs | = | complementary-determining regions |
| CL | = | clearance |
| Fab | = | fragment of antigen binding |
| Fc | = | fragment crystallizable |
| FcRn | = | neonatal Fc receptor |
| FW | = | framework region |
| hFcRn Tg32 | = | transgenic homozygous mouse model expressing human neonatal Fc receptor |
| IgG | = | immunoglobulin G |
| ITC | = | isothermal titration calorimetry |
| KD | = | dissociation constant |
| mAb | = | monoclonal antibody |
| NA | = | NeutrAvidin |
| NHP | = | non-human primate |
| pI | = | isoelectric point |
| PK | = | pharmacokinetics |
| RU | = | resonance unit |
| SA | = | streptavidin |
| SPR | = | surface plasmon resonance |
| TMDD | = | target mediated drug disposition |
| VH | = | variable heavy |
| VK | = | variable kappa |
| VL | = | variable lambda |
Introduction
Pharmacokinetics (PK) of immunoglobulin G (IgG) molecules is complex, with protective and clearance pathways that involve neonatal Fc receptor (FcRn)-dependent recycling, target-mediated drug disposition (TMDD), non-specific or off-target binding mediated clearance (CL), and sometimes immunogenicity.Citation1 Since FcRn plays an important role in modulating IgG turnover, it is hypothesized that IgG PK may correlate with binding affinity to FcRn.Citation2–19 IgG molecules can have fast CL due to TMDD, where IgG-target binding would trigger internalization and downstream degradation;Citation1 this CL pathway is not protected by FcRn. It is intriguing though that, even at high doses when all possible targets are saturated and TMDD would not be a major clearance pathway, IgG molecules with identical heavy chain subclass still have significantly varying PK.Citation20 This led us to hypothesize that differences in PK could be a result of modulation of the interaction between IgG and FcRn, most likely arising from IgG domains distal from the putative FcRn binding site. Based on crystal structures of rat FcRn complexed with rat Fc, the primary binding site of FcRn on an IgG molecule is located at the CH2-CH3 interface on the Fc domain,Citation21–24 and several studies and clinical observations have described how modulation of IgG binding affinity for human FcRn could affect IgG PK.Citation4,6,13,14 In addition to Fc modifications that enhance affinity of IgG molecules to FcRn, the existence of FcRn binding domains distal from the putative site can be an important factor that contributes to the interaction between IgG and FcRn, and thus influence PK. Several studies have suggested that the antigen binding fragment (Fab) can contribute to FcRn binding,Citation11,17–19 and a recent publication supports this hypothesis by demonstrating that the Fab region of an IgG can interact directly with FcRn.Citation16 Our hypothesis was additionally strengthened by recent publications by other groups showing a correlation between antibody charge and the FcRn affinity or PK of IgG molecules not influenced by target-mediated CL.Citation5,11,18,25,26 In the current study, we set out to further understand if Fab-containing domains can influence PK.
A detailed understanding of the FcRn-IgG interaction and a reliable, sensitive binding assay that measures this interaction would facilitate generation of an optimal monoclonal antibody (mAb) that has predictable PK in the lead optimization phase of mAb development. We set out to examine the contribution, if any, of IgG domains distal from the putative FcRn binding site on the Fc domain. Knowing that assay format can influence surface plasmon resonance (SPR) data,Citation27,28 we used a sensitive assay that measures a 1:1 interaction between IgG and FcRn to avoid avidity contributions that would complicate data analysis, as each IgG molecule can bind two FcRn molecules. Panels of IgG molecules with several combinations of variable and constant domains of heavy and light chains with sequence differences were assembled in a systematic manner, and the binding affinities between these IgG molecules and FcRn were measured using the assay to identify regions of IgG outside the structurally determined FcRn binding site on Fc that modulates affinity to FcRn.
The ability to accurately predict therapeutic antibody PK is an important factor in achieving clinical success. The PK term clearance (CL) describes the process of drug elimination from the body. For first-order elimination processes, clearance is a constant, whereas the rate of drug elimination or half-life is not constant. Volume of distribution (Vd) is another constant to describe PK. Since Vd is relatively similar across most mAbs due to their high molecular weight and limited ability to distribute to tissues, CL is a commonly used term to describe PK. PK studies in non-human primate (NHP), commonly in cynomolgus monkey, provide a reasonable and early assessment of human PKCitation29; however, NHP studies are costly and challenging to implement in the discovery and early preclinical phases of drug development, and there are ethical issues with the use of the animals. Recently, a transgenic homozygous mouse model expressing human FcRn (hFcRn, Tg32) was validated, and shown to predict human CL with 100% accuracy within 2-fold error.Citation29 In this study, PK measurements of a panel of IgG molecules were conducted in hFcRn Tg32 mice and correlation between in vitro FcRn binding and CL were assessed. Our data suggest that changes in CDRs charge can modulate affinity of IgG to FcRn in vitro. We additionally observed a correlation of FcRn affinity of a panel of IgG molecules with 1–3 mutations in CDRs only with calculated isoelectric points (pIs) of the variable regions and with CDR-L3, as well as a trend of tighter affinity associated with fast in vivo CL in human FcRn transgenic mice.
Results
Development of an SPR assay to measure FcRn-IgG binding in vitro
The goal of this study was to elucidate if IgG domains distal from the putative FcRn binding site (CH2-CH3 domain) influence binding of IgG to FcRn. SPR and isothermal titration calorimetry (ITC) were used to measure the interaction between IgG and FcRn. To reliably assess the factors governing the interaction, we believe it is necessary to preserve the natural state of an IgG molecule, including its quaternary structure and inherent flexibility in solution, to closely mimic in vivo conditions during binding experiments. For this reason, an alternative format of SPR assay was required because the existing formats either limited the flexibility of IgG by immobilizing it directly to the sensor surfaceCitation2,15 or measured avidity to FcRn that is randomly immobilized to the sensor surface at high density.Citation8,30 For this study, we developed a robust alternative SPR assayCitation31 that would allow a 1:1 interaction between FcRn and IgG. Biotinylated FcRn (through an AviTag™ on the C-terminus) was coupled to a streptavidin (SA) sensor chip allowing homogeneous orientation of FcRn. This assay format is more physiologically relevant because FcRn is present on the membrane and IgG molecules are in solution. To avoid avidity contributions that would complicate the data analysis observed at values >70 resonance units (RU) (data not shown), we tested IgG binding to FcRn coupled to the sensor chip at low density in the range 20 – 70 RU. As expected, pH-dependent binding of IgG to FcRn was observed, with binding present at pH 6.0 and absent at pH 7.4. A set of sensorgrams representing the binding of IgG (at a series of concentrations) to FcRn at pH 6.0 and dissociating at pH 7.4 is shown in . As depicted, binding using this assay format is characterized by fast association and dissociation that precluded use of available kinetic models to reliably analyze the data and obtain kinetic parameters. Instead, as rapid binding equilibrium was achieved at every concentration of IgG tested, we resorted to steady-state analysis of the data. A representative data set demonstrating the binding response as a function of the IgG concentration and the corresponding curve fit, which was used to calculate steady-state dissociation constant (KD), is shown in . Coating the sensor surface with FcRn concentrations above 70 RU resulted in tighter affinity values, poor fit of steady-state KD and non-linear Scatchard plots, indicating that avidity contributes to the affinity values with higher concentrations of FcRn on the surface (data not shown). In addition, we observed that the binding affinity of immobilized antigen-IgG complex to FcRn, where antigen was immobilized on the chip, IgG was captured and soluble FcRn was flowed over, was 3- to 96-fold lower for various IgG molecules compared to that of uncomplexed IgG to FcRn, where FcRn is captured on the chip and IgG was flowed over (), suggesting that constraining IgG molecules by binding to antigen can alter the interaction of IgG with FcRn.
Figure 1. IgG binding to FcRn in the SPR assay. A. Sensorgram representing the binding of IgG molecules (as a series of concentrations) to FcRn at pH 6.0 and dissociating at pH 7.4. The variable region from mAb-1 was cloned onto wild-type heavy chain subclass IgG2 and injected over FcRn at 0, 31.3, 62.5, 125, 250, 500, 1000, or 2000 nM. B. Steady-state KD values at equilibrium were fit using Biacore software; the affinity of mAb-1 to FcRn in this experiment was 513.4 nM.
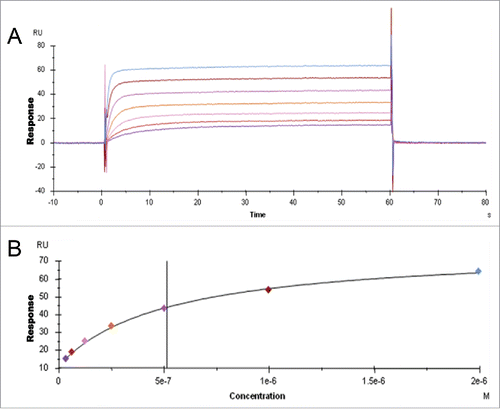
Table 1. Immobilized antigen-IgG complexes have reduced affinity for FcRn than uncomplexed IgG molecules. Steady-state KD affinity values were obtained from antigen-complexed IgG and uncomplexed IgG in SPR binding assays and values are listed for three IgG molecules.
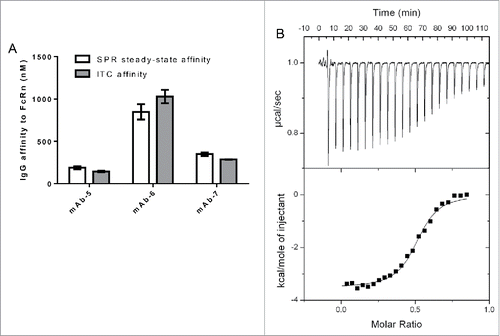
To further validate the assay format, an orthogonal technique, ITC, was used to calculate the binding affinity between three different IgG molecules and FcRn. All three IgG molecules differed in their binding affinities to FcRn; however, each of these IgG molecules exhibited similar binding affinity whether measured by SPR or ITC (). In addition, an excellent fit of ITC data to a single class of identical sites model () suggested binding stoichiometry of two identical sites for FcRn on IgG, in good agreement with previous observations.Citation32,33 The correlation between binding affinities measured using two orthogonal techniques supported the validity of the SPR assay format, which was then used to explore the IgG domains influencing the binding affinity of IgG to FcRn and their implications in vivo.
Figure 2. IgG binding to FcRn using two orthogonal techniques. A. Bar graph plotting affinity of FcRn to three IgG molecules in the SPR and ITC assays. B. ITC Trace and model fit of an IgG molecule, mAb-7, that was contained in the syringe of the ITC instrument and incrementally injected into the cell containing FcRn. Model was fit using Origin software to the one site binding model. Software generated the following: Stoichiometry (N) 0.507 ± 0.00531 Sites (IgG:FcRn), Affinity (K) 3.51e6 ± 4.90e5 M−1, change in enthalpy (ΔH) −3525 ± 51.11 cal/mol and change in entropy (ΔS) 17.9 cal/mol/deg.
Identification of IgG regions that affect affinity to FcRn
To identify domains of IgG that affect affinity to FcRn, other than the primary binding site in the Fc region, panels of IgG molecules with distinct characteristics were tested for FcRn binding using the assay described in the preceding section (). The panels included: (panel 1) IgG molecules representing subclasses IgG1, IgG2 and IgG4 bearing identical variable domains and light chain subclass to assess binding contributions of heavy chain isotype; (panel 2) IgG molecules with either kappa or lambda light chains and identical heavy chains to assess binding contributions of light chain isotype; (panel 3) IgG molecules with different variable domain frameworks (FWs) and identical heavy and (or) light constant domains to assess binding contributions of FW regions; (panel 4) IgG molecules with specificity to different target antigens and having different CDR sequences, but identical variable FW sequences and constant domains to assess binding contributions of CDRs; (panel 5) IgG molecules that bind the same antigen with 1–5 mutations in CDRs only to assess binding contributions of small changes in CDRs; and (panel 6) IgG molecules that bind the same antigen with 1–3 mutations in CDRs only to assess binding contributions of mutations that affect charge.
Table 2. Description of panels of IgG molecules tested to identify domains of IgG that affect affinity to FcRn. Steady-state KD affinity values to FcRn were determined using the SPR binding assay for panels of IgG molecules with distinct characteristics.
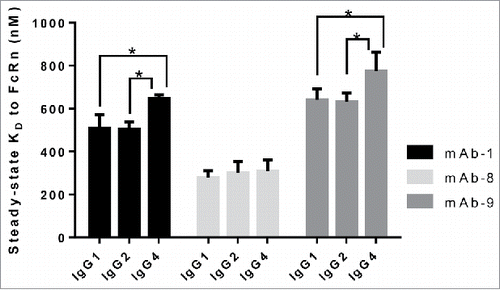
In order to determine the contribution of heavy chain subclass to the affinity of IgG to FcRn, IgG molecules (panel 1) containing variable domains derived from three mAbs were generated on subclasses IgG1, IgG2 and IgG4. All of these IgG molecules contain a kappa light chain. Three key FcRn contact residues on IgG (I253, H310, H435) are conserved in these three heavy chain subclasses, and, when mutated, were shown to ablate affinity between IgG and FcRn.Citation9 Binding affinity of wild type IgG molecules in this panel to FcRn was measured and reported in . Interestingly, the affinities of all three subclasses of mAb-8 to FcRn ranged from 279.1 to 308.2 nM, and were not statistically different from one another, indicating that changes in the heavy chain constant domains that are away from the conserved putative FcRn binding site do not alter the interaction of IgG to FcRn. Similar observations were made between IgG1 and IgG2 subclasses of mAb-1 and mAb-9. However, statistical analysis using unpaired t-test (p < 0.05) revealed that IgG4 subclass has significant differences in binding affinity compared to the IgG1 or IgG2 subclasses for mAb-1 and mAb-9. Surprisingly, differences in the binding affinity up to 2.5-fold were observed between the antibodies of similar subclass (e.g., IgG1 versions of mAb-8, mAb-1 and mAb-9), suggesting an influence of changes in variable domains on the affinity of IgG to FcRn. This is in agreement with recent observations that the Fab domain of monoclonal IgG molecules affects IgG interaction with FcRn,Citation11,16,18,19 thus prompting us to further investigate the role of various structural elements of Fab domains on the IgG interaction with FcRn.
Figure 3. Heavy chain subclass does not alter the interaction of IgG to FcRn. IgG molecules from panel 1, with differing heavy chain subclasses, were tested for affinity to human FcRn in the SPR assay a minimum of three times on three different surfaces in three separate experiments. Mean steady-state KD values for each IgG molecule were plotted with error bars indicating standard deviations. Each set of IgG molecules, containing the same variable sequence and light chain subclass, is colored differently. Significant steady-state KD differences between IgG molecules within the same set were analyzed using an unpaired student t-test where significance is indicated as single asterisk (*) for p < 0.05.
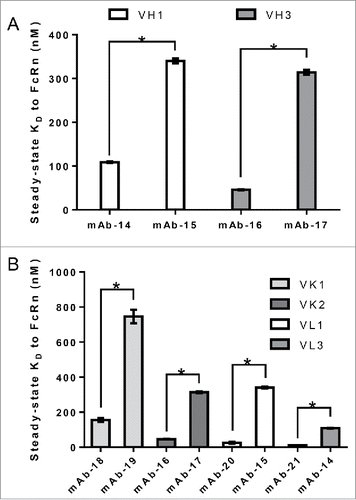
The Fab region constitutes both variable and constant domains of heavy and light chains. Panel 2 represents a set of IgG molecules that differ in the light chain subclass and contain identical heavy chain subclass (IgG1). As shown in , a wide range of FcRn binding affinity values were observed for all of the mAbs within each kappa and lambda group; kappa ranging from 10.7 to 811 nM and lambda ranging from 12.6 to 848 nM. Statistical analysis using unpaired t-test (p < 0.05) revealed significant differences in binding affinity for the mAbs within each kappa and lambda group. In addition, two versions of an antibody with identical target binding affinities, consisting of identical variable sequences and heavy chain isotype, but differing constant light chain domains (one kappa and the other lambda) bound to FcRn with similar affinity (data not shown). This observation, together with those described in , strengthened the hypothesis that variable domains, which contain domains from both heavy and light chains, could modulate binding affinity to FcRn. Variable domains of both heavy and light chains can be further divided into FWs and CDRs that are anchored by FWs. In order to determine the contribution of variable domain FWs to the affinity of IgG to FcRn, panel 3 mAbs with different heavy and light chain variable domain FW families and identical corresponding heavy and light chain subclasses were compiled and tested for binding to FcRn. As shown in , statistical analysis using unpaired t-test (p < 0.05) revealed IgG molecules bearing identical variable heavy (VH) FWs bound FcRn with significantly differing affinity; the VH1 family showed affinity values of 108.8 and 340.1 nM, and the VH3 family showed affinity values of 45.9 and 314.1 nM. Comparable observations have been made for antibodies bearing identical variable kappa (VK) or variable lambda (VL) FWs, suggesting that VH chain FW may not contribute to the interaction of IgG to FcRn (). Because no trend between affinity of IgG to FcRn and the variable domain FWs was observed, we further hypothesized that CDRs might play a role in modulating affinity to FcRn.
Figure 4. Light chain subclass does not alter the interaction of IgG to FcRn. IgG molecules from panel 2, containing kappa or lambda light chains all paired with the same heavy chain subclass (IgG1), was compiled and tested for affinity to FcRn in the SPR assay a minimum of two times in duplicate on two different surfaces in two separate experiments. The mean steady-state KD values for each IgG molecule were plotted with error bars representing standard deviations. Kappa and lambda containing IgG molecules are colored differently. Significant steady-state KD differences between IgG molecules with the same light chain isotype were analyzed using an unpaired student t-test where significance is indicated as single asterisk (*) for p < 0.05.
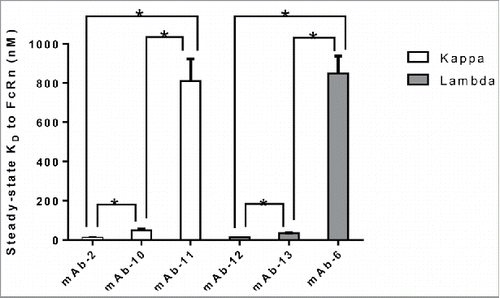
Figure 5. Variable domain FW family does not alter the interaction of IgG to FcRn. IgG molecules from panel 3, containing different variable domain FW families all paired with identical heavy and light chain subclasses, were compiled and tested for affinity to FcRn in the SPR assay a minimum of two times in duplicate on two different surfaces in two separate experiments. Mean steady-state KD values for each IgG molecule was plotted with error bars representing standard deviations. Different FW family containing IgG molecules are colored differently. Significant steady-state KD differences between IgG molecules with the same FW family were analyzed using an unpaired student t-test where significance is indicated as single asterisk (*) for p < 0.05. A. Heavy chain variable FW domain families VH1 and VH3. B. Light chain variable FW domain families VK1 and VK2, and VL1 and VL3.
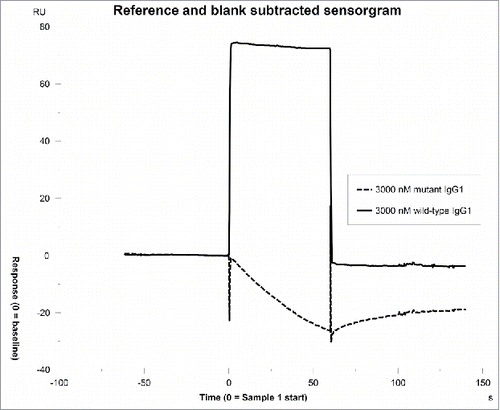
To test the influence of CDRs on the IgG affinity to FcRn, six IgG molecules (panel 4) containing identical constant domains and FW residues, but different CDRs with specificity to different targets were assessed for binding to FcRn. The six IgG molecules exhibited affinities to FcRn in the range of 49.3–1318 nM, indicating that CDR sequences can alter the affinity of IgG to FcRn (). To validate this observation, a different set of 29 IgG molecules with a small number of mutations in CDRs only was compiled (panel 5) and are described in . IgG molecules in this set were derived from the same parent IgG molecule, but had tighter binding affinity to antigen relative to the parent. In any given IgG within the set, 1–5 mutations in CDRs exist. The IgG molecules exhibited affinities to FcRn in the range of 3.2–284.7 nM, indicating that even small changes in CDR residues of an IgG molecule can dramatically alter the binding affinity of IgG to FcRn (). Intrigued by this observation, we queried if the Fab domain alone could bind to FcRn in the absence of binding at the putative site on the Fc domain. To address this question, IgG molecules that exhibited higher affinities in the range of 15.4–104.3 nM to FcRn were mutated at the two key histidine contact residues (H310A, H435A). These mutations have been shown to ablate the interaction between IgG and FcRn.Citation7,34 A representative sensorgram resulting from binding of mAb-10 to FcRn is presented in . As shown, no interaction was measured between IgG mutant and FcRn even at IgG mutant concentration as high as 3000 nM (a concentration value just above 60-fold the steady-state KD value of the wild type IgG molecule). Based on this observation, we concluded that the primary interaction between the Fc that constitutes the putative binding site and FcRn is absolutely necessary, and then minor contributions from CDRs can further strengthen the interaction. It is possible that residues in CDRs could directly contact FcRn following the first interaction at the putative binding site, owing to the inherent flexibility of Fab arms.Citation16,35-37
Figure 6. CDR sequences can alter the binding affinity of IgG to FcRn. Mean steady-state KD values for each IgG molecule in panel 4 were plotted with error bars representing standard deviations. Each IgG molecule contains identical constant domains and FW residues, but different CDR sequences.
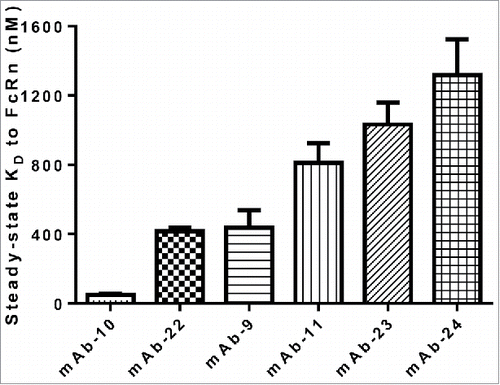
Table 3. Small variations in CDRs alter affinity of IgG to FcRn. IgG molecules in panel 5, differing only by 1–5 mutations in CDR sequences, were tested for affinity to FcRn in the SPR assay.
Figure 7. No binding is observed between the Fab domain and FcRn. Variable domains from mAb-10 were cloned onto IgG1 subclass with mutations in the two key histidine contact residues to ablate the interaction between the Fc regions of IgG and FcRn. No interaction was measured after injecting these IgG molecules at maximum concentrations just over sixty times the steady-state KD of the wild type IgG1 molecule.
Correlation between pI of various domains of IgG and affinity to FcRn
The observations described in the preceding paragraphs along with the published results that demonstrated the effect of charges in antibody FWs and CDRs on the in vivo half-lifeCitation5,18,25,26 encouraged us to hypothesize that charges in CDR domains may directly or indirectly influence IgG affinity to FcRn. To test the hypothesis, theoretical pI values were calculated for various domains of IgG molecules from panel 4, which differ in CDRs only. The calculated pI values and mean steady-state KD of the interaction between FcRn and each IgG molecule tested are reported in Table S1. As shown in , variable domain pI correlated very well with IgG affinity to FcRn (r = −0.85, p < 0.05), indicating that charge modulation in the CDRs of variable domains can affect the FcRn binding affinity. Further analysis of IgG binding affinity in relation to individual CDR pI revealed good correlation with changes in CDR-L1 (r = −0.82) and L3 (r = −0.88), but not with changes in CDR-L2, H1, H2 or H3. To further validate the correlation of IgG binding affinity with pI of CDR-L3, a subset of five of the IgG molecules in panel 5 that differ by mutation of a single amino acid in CDR-L3 only was analyzed (mAb-25, 26, 27, 38 and 42). An increase in the pI of variable region and CDR-L3 correlated (r = −0.99 and −0.91, respectively), with tighter binding affinity of IgG to FcRn (Table S2 and ).
Figure 8. Correlation of various pI values with affinity to FcRn. pI values were calculated for the entire IgG, variable regions and each individual CDR for the IgG molecules in panel 4, which differ by CDRs only. pI values of variable regions (r = −0.85), CDR- L1 (r = −0.82) and L3 (r =−0.88) all correlate with their corresponding affinity to FcRn. No correlation was observed with pI of CDR-L2 (r =−0.53), H1 (r =−0.25), H2 (r =−0.66) or H3 (r =−0.70).
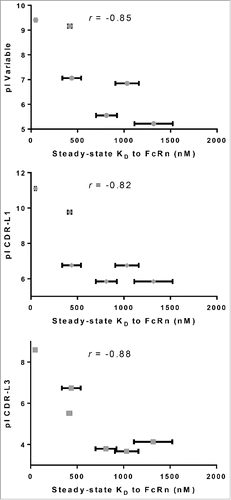
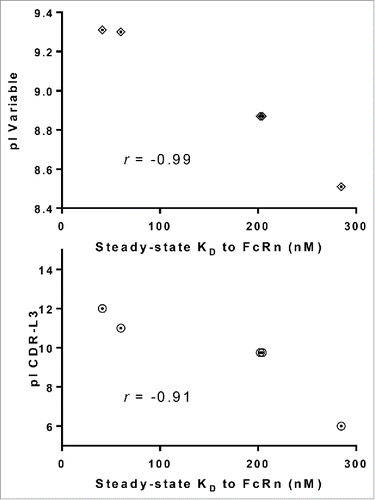
Figure 9. Higher pI values of CDR-L3 correlates with higher affinity to FcRn. Plotting the steady-state KD vs. pI of variable regions and CDR-L3 for a subset of IgG molecules from panel 5 that differ by only one amino acid residue in CDR-L3 (mAb-25, 26, 27, 38 and 42) demonstrated a strong correlation between higher pI value and higher affinity to FcRn for variable regions (r =−0.99) and CDR-L3 (r =−0.91).
Based on these observations and our understanding of the contribution of CDRs to modifying affinity to FcRn, a structural model was generated to examine whether CDRs in the Fab arm of an IgG might directly contact FcRn (). After examining the model and observing the proximity of Fab arm to the endosomal membrane, we hypothesized that flexibility in the hinge of IgG may allow for the Fab arm to contact either FcRn directly and (or) the membrane following binding to FcRn. We believe that our SPR assay format allows mimicking of the membrane containing FcRn owing to the extremely flexible and negatively charged nature of the SPR chip matrix. To test the possibility of the Fab arm contacting the membrane, we examined the binding affinity of a panel of five IgG molecules varying only by 1–3 mutations in CDRs (panel 6) to FcRn in the presence and absence of the carboxymethyl dextran matrix. NeutrAvidin (NA), a neutrally charged version of SA, was coupled to a matrix-free surface. Biotinylated FcRn was coupled to the resulting sensor surface and binding experiments were carried out. All these IgG molecules bind the same antigen and the mutations in CDRs do not alter affinity to antigen. Each IgG molecule has one mutation in the middle of CDR-L3 that is positively charged in mAb-54 and −57, neutral in mAb-55 and 56 and negatively charged in mAb-58. The other one or two mutations, when present, are in CDR-H1 or H2. As reported in , the affinity of these IgG molecules to FcRn ranged from 101.3–682.1 nM in the assay with no carboxymethyl dextran matrix. To confirm that this difference is indeed due to the absence of the matrix and not due to the substitution of SA with NA, we coated NA on the surface of a CM3 chip, a matrix most closely resembled to that in the SA chip, captured biotinylated FcRn on this sensor and measured the affinity of this set of IgG molecules. The affinities were in the range of 15.0–674.1 nM, which is similar to that observed with the SA-coated chip (), indicating minimal difference between SA- or NA-based matrices. Statistical analysis using unpaired t-test (p < 0.05) revealed that removal of the matrix significantly weakened the affinity of mAb-54, 55, 56 and 57 to FcRn, but not mAb-58, suggesting the positive charges in the CDRs of these IgG molecules may be interacting with the negatively charged matrix. To understand if the flexibility of Fab arms would affect IgG binding to FcRn, we coated the surface of a CM5 chip with the cognate antigen, captured a low density of the antibody so the Fab arms are constrained and flowed FcRn over to measure the binding affinity of FcRn. In this format the affinity values are much weaker, ranging only from 1518–1850 nM, suggesting that IgG flexibility can alter the interaction of IgG with FcRn quite significantly and that the CDRs of IgG molecules in panel 6 influence affinity to FcRn when IgG exists in a non-constrained form, which represents the physiological state. For this set of IgG molecules, the variable domain pI and CDR-L3 pI correlated with IgG affinity to FcRn in the standard SA assay (r =−0.96 and−0.92, respectively), indicating that charge modulation in the CDRs of variable domains can affect the FcRn binding affinity.
Figure 10. Model of IgG binding to FcRn. FcRn is a heterodimer consisting of a MHC-class-I-like heavy chain, FcRn (shown in green) and a β2 m light chain (shown in red). The transmembrane domain is drawn in through a cartoon of the endosomal membrane. The figure displayed is modeled on the crystal structure of rat FcRn binding rat IgG2a Fc (PDB ID: 1FRT11) superimposed with modeled IgG1 based on the crystal structure of IgG1 (PDB ID: 1HZH). In this model, the right side of the Fc of IgG1 heavy chain (shown in grey) is binding to FcRn. One of the Fab arms (shown in yellow with CDRs in pink) is facing towards the endosomal membrane.
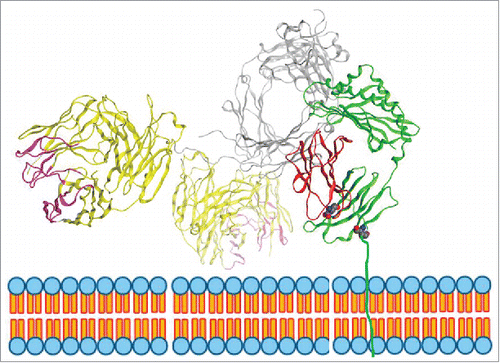
Figure 11. Presence of the carboxymethyl dextran matrix and constraining the Fab domain of IgG molecules by binding to antigen alters steady-state KD to FcRn. The FcRn SPR binding assay was carried out on SA and NA chips with a carboxymethyl dextran matrix, on NA coated on a C1 chip with no matrix and on IgG molecules captured on antigen for the five IgG molecules from panel 6 that vary by 1–3 amino acid residues in CDRs only on at least three experiments on a minimum of two different surfaces. Significant steady-state KD differences between IgG molecules tested with or without matrix were analyzed using an unpaired student t-test where significance is indicated as single asterisk (*) for p < 0.05.
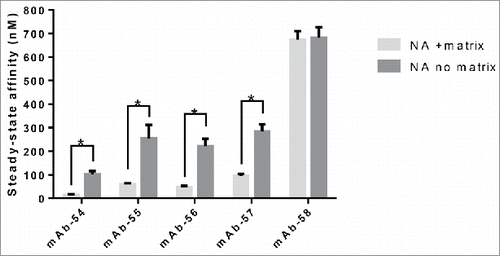
Table 4. CL values for the set of IgG molecules from panel 6, which differ by 1–3 amino acid residues in CDRs only, show big variation in CL (mL/hr/kg). hFcRn Tg32 homozygous mice were IV dosed with 5 mg/kg of mAb. For mAb-54, the last two timepoints were excluded from PK parameter calculation due to presumed TMDD or ADA. In vitro affinity to FcRn, location and number of mutations, and pI values of variable and CDR-L3 are also listed for each IgG molecule.
Relationship between in vitro IgG affinity to FcRn and in vivo clearance
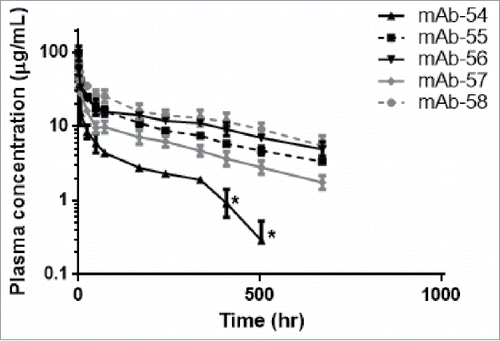
In order to understand whether the affinity values of IgG to FcRn relate to the observed in vivo CL, we administered the five IgG molecules in panel 6 to hFcRn Tg32 homozygous mice, which have been shown to accurately predict mAb CL in humans.Citation29 As previously noted, all the IgG molecules in this panel bind to the same antigen and vary only by 1–3 mutations in their CDRs; all five contain a single amino acid mutation in the middle of the CDR-L3 loop. The mutations in CDRs do not alter affinity to antigen, but introduce differences in charge between the IgG molecules. Furthermore, these IgG molecules do not cross-react to mouse antigen, eliminating the possibility of TMDD contribution to CL. Following a 5 mg/kg intravenous administration, the IgG molecules displayed variable exposures in the hFcRn Tg32 mouse model, resulting in a range of CL values from 0.42–2.70 mL/hr/kg (). mAb-54 had the fastest CL and tightest affinity to FcRn while mAb-58 had the slowest CL and weakest affinity to FcRn (). mAb-54 contains a positively charged amino acid in CDR-L3 while mAb-58 contains a negatively charged amino acid in place of the positively charged residue in CDR-L3. These data suggest that charges in CDRs can affect the resulting in vivo CL.
Figure 12. Mutations in CDRs can alter in vivo CL of IgG molecules. Plasma concentrations from hFcRn Tg32 homozygous mice were plotted over time for IgG molecules from panel 6, a set of five IgG molecules that vary by 1–3 amino acid residues in CDRs only. For mAb-54, the asterisks (*) indicate timepoints excluded from PK parameter calculation due to presumed TMDD or anti-drug antibody (ADA).
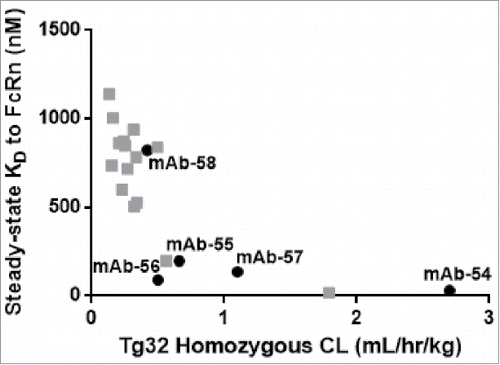
To further examine the relationship between in vitro affinity of IgG molecules to FcRn and in vivo CL, we used a data set generated previously that included in vivo CL and in vitro affinity values for 27 additional IgG molecules,Citation29 including those from panel 6. As shown in , FcRn steady-state KD values correlated (r =−0.79) with in vivo CL in the hFcRn Tg32 homozygous mice, demonstrating that IgG molecules with tighter in vitro affinity have fast in vivo CL.
Figure 13. In vitro affinity to FcRn correlates with in vivo CL of IgG molecules. Plotting the hFcRn Tg32 homozygous CL vs. the steady-state KD for the set of five IgG molecules from panel 6 that vary by 1–3 amino acid residues in CDRs only (mAb-54–58, in black) and twenty-one additional IgG molecules (gray) demonstrated a correlation between in vitro affinity to FcRn and in vivo CL (r =−0.79).
Discussion
A substantial body of literature exists demonstrating the role of FcRn in extending the PK of therapeutic antibodies by rescuing them from lysosomal degradation. Although all the therapeutic antibodies currently marketed are of the IgG isotype, they differ substantially in their in vivo PK. In addition to FcRn binding affinity, a factor that often influences the PK of IgG molecules is TMDD. However, studies have shown that, even in the absence of TMDD, PK of IgG molecules can differ significantly, indicating that other factors might be playing a role. For example, specific off-target binding of a humanized anti-fibroblast growth factor receptor 4 IgG to mouse complement component 3 exhibited rapid CL in mice.Citation38 Non-specific binding of IgG may also result in poor serum PK, as was seen with variants of palivizumab, which targets respiratory syncytial virus.Citation39 Several studies have demonstrated that PK can be manipulated by modulating the binding affinity between IgG and FcRn through engineering of the Fc region of IgG. Introducing mutations that enhance FcRn binding at endosomal pH, where IgG is exposed to FcRn, but not at neutral pH, is thought to allow more IgG to dissociate from FcRn after fusion of the endosome with plasma membrane as the half-lives of IgG molecules with these mutations are enhanced compared to the same IgG molecules with wild type FcRn contact residues.Citation4,6,13,14 In our study, we sought to understand the role of different IgG domains that are distal from the putative binding site in modulating the affinity between IgG and FcRn.
Therapeutic IgG antibodies are of different subclasses and are multi-domain proteins, each domain serving a distinct function. A study measuring human maternal-fetal transport of IgG showed differences in transport for the four heavy chain subclasses, in the order of IgG1 > IgG4 > IgG3 > IgG2,Citation40 and, based on this study, it has been widely assumed that FcRn recycling is most effective with the IgG1 subclass. In contrast to this widely accepted hypothesis, we demonstrated that the affinity of IgG with identical variable domains to FcRn is similar for IgG1, IgG2 and IgG4 subclasses (). It has been demonstrated that the IgG4 hinge has enhanced flexibility compared with that of IgG1,Citation37,41 and that the Fab domain of IgG4 molecules can restrict access to the Fc region.Citation42 Therefore, it is possible that, in the case of IgG4, additional hinge flexibility slightly alters its interaction with FcRn, and may account for the small reduction in binding affinity we observed (). While IgG subclass showed no significant effect on FcRn binding affinity, surprisingly, the Fab domain significantly affected binding affinity, as exemplified by the FcRn binding of three different antibodies of identical subclass (). Additionally, we observed that IgG molecules immobilized as antigen-IgG complexes have reduced affinity for soluble FcRn compared to uncomplexed IgG molecules flowed over immobilized FcRn (). Similar lower affinity values to FcRn were also observed by Igawa et al.Citation25 in an SPR assay where IgG molecules were immobilized on anti-kappa antibodies and soluble FcRn was used as an analyte. Together, these observations suggest that constraining the flexibility of IgG reduces affinity to FcRn. Another possibility for reduced affinity could be attributed to the ability of IgG molecules to form higher order non-covalent structures such as hexamers. Such structures have been shown to exist under high antigen densities found for example on the surface of viral particles that are presented to complement system by the antibodies binding to these surface antigens.Citation43,44 However, we believe such structures may not exist under the SPR conditions used in the present study as the FcRn density on the chip was calculated to be low, in the order of 0.5 ng/mm2, and the IgG molecules examined have high affinity to antigen with slow dissociation rates. It was shown by Hadzhieva et al.Citation45 that immobilization of antigen at such low densities has not resulted in higher order antibody structures. Structural changes in IgG molecules as a result of antigen binding have been reported by other groups as well,Citation46 substantiating our observations. Encouraged by these observations, we sought to systematically dissect the contributions of variable domain in modulating the affinity between FcRn and IgG.
The variable domain is composed of FWs that form the core and CDRs or paratopes that confer specific binding to the cognate antigen. This study demonstrates that the FW domains comprising both heavy and light chains did not affect FcRn affinity to IgG molecules, as wide ranges of affinity values were measured for IgG molecules containing identical FW domains (). The next logical step was to evaluate the contribution of CDRs to FcRn affinity. The binding affinity of IgG molecules in panels 5 and 6 to FcRn that differed by only a few amino acid residues in CDRs revealed that small changes in CDRs, as minute as one amino acid residue change, could alter affinity to FcRn up to 79-fold ( and ). These observations are intriguing because CDR domains are distant from the putative FcRn binding site on the Fc. This led us to question whether CDRs could directly contact FcRn or the nearby membrane, influence flexibility of the Fab arm, or influence the tertiary or quaternary structure of IgG, and thus modulate affinity of IgG to FcRn. It is generally surmised that individual IgG molecules are highly flexible; allowing Fab domains to come in close proximity to Fc. Inherent flexibility of IgG increases the likelihood of its contact with the cognate antigen. The Fab arm can rotate as much as 158°, and the Fab-Fc angles ranged from 63–172° as revealed by electron tomography studiesCitation35,36 and the existing crystal structures. Such flexibility might increase the likelihood of variable domains initiating contact with FcRn, following FcRn binding at its primary binding site on the Fc domain. illustrates a model of human FcRn in complex with human IgG constructed based on the crystal structure of rat Fc-FcRn and demonstrates the possibility of direct contact between Fab arm and FcRn, specifically with a region of the beta-2 microglobulin (β2m) subunit with negatively charged surface patches. Recent work by Jensen et al.Citation16,47 demonstrated that there are interaction sites in the Fab region of IgG for FcRn, which corroborates our hypothesis that the Fab arm may directly contact FcRn.
The other likely biological surface the Fab region may contact following its binding to FcRn is the membrane where FcRn is presented. This has been suggested as a possibility by Burmeister et al.Citation23 based on the rat Fc-FcRn crystal structure and modeling. Further evaluation of the structural model suggests that the Fab domain might likely come in contact with the endosomal/vesicle membrane. Findings by Wang et al.Citation19 demonstrated that the Fab domain affected the interaction with FcRn and that in vitro FcRn dissociation at neutral pH correlated with in vivo half-life. Additionally, Jensen et al.Citation16,47 demonstrated FcRn interaction sites in the Fab region of IgG. However, under the SPR conditions we tested, a direct interaction between FcRn and Fab domain has not been observed in the absence of primary interaction at Fc domain (). We reasoned that this interaction may not be as strong or specific enough to be detected in our SPR assay. Nevertheless, this weak interaction might securely position the FcRn molecule and strengthen the primary interaction at the putative site.
Based on these assumptions, we hypothesized that modifying charges in CDRs might modulate this secondary contact. Calculated pI of variable domains from IgG molecules from panel 4, which differ by CDRs only, ranged from 5.22–9.41 (Table S1). We found that the higher the pI, the tighter the binding affinity (). Further dissection of the variable domain pI showed that higher pI of CDR domains L1 and L3 correlated with tighter affinity to FcRn, with L3 showing the strongest correlation (). Furthermore, the binding affinities of a subset of five IgG molecules from panel 5, differing by only one amino acid substitution in CDR-L3, showed that higher pI of variable and CDR-L3 domains correlated with tighter affinity to FcRn (). This suggests that charge modulation in the CDRs of variable domains may affect the FcRn binding affinity. It should be noted that the specific CDR domains that influence FcRn binding as a result of charge modulations may differ from one IgG molecule to another. We also examined the influence of the negatively charged carboxymethyl dextran matrix that constitute the SPR chips and likely mimic the negatively charged plasma membrane, and observed that the presence of the matrix alters interaction of some IgG molecules to FcRn, but not others (). Depending on local charges in the IgG molecule, it is possible that the matrix and (or) regions of FcRn distal from the putative binding site may attract or repel the IgG, and thus alter its affinity to FcRn.
While several factors can influence the PK of an IgG molecule, FcRn-dependent recycling is one of the key factors. Published data correlating the in vitro affinity of IgG molecules for FcRn and PK parameters does not support a linear relationship between affinity and half-life.Citation30,48,49 In order to measure the interaction between IgG and FcRn, we believe it is highly desirable to preserve the physiologically relevant quaternary structure and inherent flexibility in solution during binding experiments, and this is captured in the SPR assay format that we described here. We believe that this format helped us draw correlations between the in vitro affinity of IgG molecules to FcRn and IgG CL. Our data reported in demonstrates that IgG molecules with in vitro affinity tighter than 400 nM often have rapid in vivo CL, suggesting the likely contributions from CDRs in the Fab arm and likely slow release of the IgG when recycling. In contrast, IgG molecules with weaker affinity than 400 nM fall into a category of slower CL, possibly due to other in vivo factors that more likely modulate IgG half-life. In addition, we report that the relationship between the in vitro affinity between IgG and FcRn and IgG CL may not be linear for IgG molecules with affinity weaker than 400 nM. Based on the reported in vivo study results and in vitro observations, our working hypothesis is that IgG molecules with tighter affinity to FcRn as a result of contributions, particularly from CDRs in the Fab arm, may not rapidly dissociate from FcRn at the cell surface during the “kiss-and-run” fusion, and are instead re-internalized with FcRn. Engineering CDRs to modify local charge and thus maintain affinity to FcRn at 400 nM or weaker in vitro while retaining antigen binding may have far-reaching implications in the half-life optimization efforts of IgG therapeutics with respect to in vivo PK.
Materials and methods
Generation of biotinylated human FcRn- β2 m complex
DNA representing the extracellular domain of FcRn, including the signal peptide, (amino acid residues 1–290; accession no. BC008734), was amplified from cDNA (Open Biosystems) using PCR. The PCR fragments were further extended on the 5′ end to add a short linker sequence (GGGSGGG) followed by AviTag™ (Avidity, LLC) (GLNDIFEAQKIEWHE) using overlapping PCR. The resulting construct was cloned into a proprietary expression vector. β2 microglobulin (β2m) cDNA (Ultimate ORF clone, Invitrogen) was amplified using PCR (amino acids 1–119) and the resulting product was cloned into a proprietary expression vector. Both FcRn and β2m clones were co-transfected into Chinese hamster ovary (CHO) cells and clones were selected by growing them in growth medium containing 20, 50 or 100 nM MTX and 1 ug/mL G418 to create stable cell lines. Clones from each MTX concentration were isolated, and cultured in growth medium. Supernatants were analyzed for protein expression on 4–12% NuPAGE gels (Invitrogen), followed by Western blot using goat polyclonal anti-FcRn antibody (Santa Cruz Biotechnology; sc-46329) and mouse monoclonal anti-β2m antibody (Santa Cruz Biotechnology; sc-15366). The best expressing clonal lead cell line was selected for scale-up.
Stable CHO lines expressing FcRn were seeded into roller bottles (Cellon) at 2.7e7 cells/roller bottle. Four days post seeding, the serum containing growth media was removed, the roller bottles were rinsed once with 200 mL of phosphate-buffered saline (PBS), and 400-mL/roller bottle of a proprietary serum-free media was added. Seventy-two hours after changing to serum-free media, the conditioned media was removed and pooled. Immediately after harvesting, the pooled conditioned media was clarified by filtration and concentrated ∼ 20 fold using a 10 kD tangential flow filtration cartridge (Millipore Prep Scale Cartridge). The pH of the harvested conditioned medium containing FcRn was adjusted to 5.5–6.0 with 1 M 2-(N-morpholino)ethanesulfonic acid (MES), pH 5.0, and captured on a column containing IgG-coupled sepharose6 fast flow resin (GE Healthcare). Following an extensive wash, FcRn- β2m heterodimer was eluted in PBS-calcium/magnesium free, pH 7.5.
Purified FcRn was incubated with biotin ligase and biotin to site-specifically attach biotin to the lysine residue in the AviTag™ following manufacturer's instructions (GeneCopoeia). Biotinylation was confirmed by Western blot analysis using streptavidin-horseradish peroxidase (SouthernBiotech) for detection.
Generation of IgG molecules
Monoclonal IgG molecules used in this study were discovered using both phage display and hybridoma approaches. PCR site-directed mutagenesis was used to introduce mutations into the Fc portion of human IgG. All mAbs used in this study were produced as recombinant proteins using either human embryonic kidney (HEK293) cells or CHO cells and purified by Protein A resin (GE Healthcare). All pure mAbs were determined to be >99% aggregate-free by analytical size exclusion chromatography.
SPR FcRn-IgG binding assays
Biacore™ T100 or T200 (GE Healthcare) were used to measure steady-state binding affinities of the interaction between IgG molecules and FcRn. Biotinylated human FcRn-β2m complex was affinity captured on the surface of a SA chip (GE Healthcare) to a final surface density of 20–70 RU. IgG analytes were diluted to appropriate experimental concentrations in sample buffer (20 mM MES, 150 nM NaCl, 3 mM EDTA, 0.5% P20, pH 6.0) and flowed over the FcRn-coated SA chip. Following the association and dissociation phases of the experiment, the SA chip was regenerated using 30 s injection of 10 mM HEPES with 150 nM NaCl, 3 mM EDTA, 0.05% P20, pH 7.4 (HBS-EP+ buffer, GE Healthcare) prior to the next cycle. Unless otherwise mentioned, assays were performed in triplicate independently using a minimum of three different FcRn-coated SA chips and the average steady-state KD value and standard deviation for each IgG molecule were calculated. IgG molecules described in , Table S2 and were analyzed in only a single experiment due to the limited quantities of protein available. Appropriate control experiments were carried out to show that mass transport was minimal and the interaction was truly a 1:1 binding event. The resulting sensorgrams were double referenced and fit to steady-state affinity models using the Biacore T200 Evaluation Software (version: 1.0, GE Healthcare) or Biacore T100 Evaluation Software (version 2.0.1, GE Healthcare). The validity of the data was assessed using calculated chi-square to measure goodness of fit to the binding model and Rmax values to verify that all FcRn and IgG molecules are actively binding.
Antigen, specific for mAb-2–4 and mAb-54–58, was coated by amine coupling on the surface of a CM5 chip (GE Healthcare). mAb-2–4 were diluted into HBS-EP+ buffer and were affinity captured on the antigen. Human FcRn-β2m complex was diluted to appropriate experimental concentrations in sample buffer (20 mM MES, 150 nM NaCl, 3 mM EDTA, 0.5% P20, pH 6.0) and flowed over the IgG captured by its antigen on the CM5 chip. Assays were performed in duplicate independently using two different antigen-coated CM5 chips and the average steady-state KD value and standard deviation for each IgG molecule were calculated. The resulting sensorgrams were double referenced and fit to steady-state affinity models using the Biacore T200 Evaluation Software (version: 1.0, GE Healthcare) or Biacore T100 Evaluation Software (version 2.0.1, GE Healthcare). The validity of the data was assessed using calculated chi-square to measure goodness of fit to the binding model and Rmax values to verify that all FcRn and IgG molecules are actively binding.
50 ug/ml NeutrAvidin (Thermo Scientific), in 10 mM sodium acetate, pH 4.0, was injected over all four flow cells on a C3 chip (GE Healthcare) to coat all flow cells with NA. Biotinylated human FcRn-β2m complex was affinity captured on the surface of an SA chip (GE Healthcare) to a final surface density of 40–50 RU. IgG analytes were diluted to appropriate experimental concentrations in sample buffer (20 mM MES, 150 nM NaCl, 3 mM EDTA, 0.5% P20, pH 6.0) and flowed over the FcRn-coated NA chip. Following the association and dissociation phases of the experiment, the NA chip was regenerated using 30 s injection of 10 mM HEPES with 150 nM NaCl, 3 mM EDTA, 0.05% P20, pH 7.4 (HBS-EP+ buffer, GE Healthcare) prior to the next cycle. Assays were performed in triplicate independently using a minimum of three different FcRn-coated NA chips and the average steady-state KD value and standard deviation for each IgG molecule were calculated.
50 ug/ml NA (Thermo Scientific), in 10 mM sodium acetate, pH 5.0, was injected over all four flow cells on a C1 chip (GE Healthcare) using C1 pre-concentration buffer (100 mM glycine, pH 12 with 0.03% TritonX100) to coat all flow cells with NA. Biotinylated human FcRn- β2m complex was affinity captured on the NA on flow cells 2–4 to a final surface density of 11.5–40 RU. IgG molecules were diluted to appropriate experimental concentrations in sample buffer (20 mM MES, 150 nM sodium chloride, 3 mM ethylenediaminetetraacetic acid, 0.5% P20, pH 6.0) and flowed over the FcRn-coated NA chip. Following the association and dissociation phases of the experiment, the chip was regenerated using 30 s injection of 10 mM HEPES with 150 nM sodium chloride, 3 mM ethylenediaminetetraacetic acid, 0.05% P20, pH 7.4 (HBS-EP+ buffer, GE Healthcare) prior to the next cycle. Assays were performed in triplicate independently using three different FcRn-coated NA chips and the mean steady-state KD value and standard deviation for each IgG molecule were calculated. The resulting sensorgrams were double referenced and fit to steady-state affinity models using the Biacore T200 Evaluation Software (version: 1.0, GE Healthcare). The validity of the data was assessed using calculated chi-square to measure goodness of fit to the binding model and Rmax values to verify that all FcRn and IgG molecules are actively binding.
Isothermal titration calorimetry affinity measurements
ITC experiments were carried out using a VP-ITC instrument (GE Healthcare). All experiments were performed with FcRn in the reactant cell and IgG in the titrating syringe. Both FcRn and IgG were extensively dialyzed in 50 mM sodium phosphate, 150 mM sodium chloride, pH 6.0 and the same buffer was used in reference cell. Concentrations of FcRn and IgG were adjusted to 0.014 mM or 0.028 mM (FcRn) and 0.11 mM (IgG) using the buffer mentioned above and degassed prior to adding to the reactant cell and the titrating syringe. All runs were performed at 20°C, and each run comprised a series of 22 injections of 13.3 µL IgG each with syringe stir speed of 470 rpm. The injections were spaced 300 s apart to allow time to reach equilibrium prior to the next injection. Data were analyzed using Origin 7.0 (Origin Lab) and fit to the one site binding model to calculate stoichiometry and binding affinity.
Isoelectric point determination
Isoelectric point (pI) values of variable domains and CDRs were deduced from amino acid sequence using ExPASy (Swiss Institute of Bioinfomatics).
Statistical analysis
A Student's unpaired t-test analysis was performed to compare steady-state KD values between IgG molecules. Differences were considered as statistically significant if p < 0.05 (labeled as *).
Analysis of correlations between IgG molecule steady-state KD value to calculated pI value of variable domains and CDRs was performed using GraphPad Prism 6 to determine the Pearson correlation coefficient (r) where statistical significance level is set at or below 0.05. All correlations were fit using a linear regression fit.
PK studies conducted in hFcRn Tg32 mice
Mice used in this study included hFcRn Tg32 strain (cat#014565, homozygous) obtained from The Jackson Laboratory. All mice were treatment-naïve male mice between the ages of 6 to 8 weeks at study start. PK studies were conducted at Pfizer Inc., and were designed and executed within accordance of the Animal Use Protocol and adherence to the Pfizer Institutional Animal Care and Use Committee regulations. mAb dosing solutions were prepared in a 10 mM L-histidine, 5% sucrose, pH 6.0 formulation buffer and dosed intravenously at 5 mg/kg with a dose volume of 4 mL/kg. A total of 6 animal replicates were evaluated for each mAb utilizing a serial sampling approach as previously describedCitation50 across a study duration of 4 weeks. Serum concentrations of mAb were determined using a generic human IgG ligand binding assay (LBA) as described below. PK parameters were determined from individual animal data using non-compartmental analysis in WinNonlin (Version 6.3.0.395) using the plasma data module. Terminal data points in PK profiles that were presumed to be affected by TMDD, off-target binding or anti-drug antibody (ADA) were excluded from the linear CL estimation (336–504 hr from mAb-54 only).
Serum quantitation of mAb by LBA
mAb serum concentrations were quantitated as previously described.Citation29,50 Briefly, samples were collected via serial sampling, 10 μL of whole blood was collected at each time point via a heparinized capillary tube and mixed with 90 μL of assay buffer (0.2 M Tricine, pH 8.5, 1% bovine serum albumin (BSA), 0.5 M NaCl, 0.1% Zwittergent, 0.05% Proclin300) to obtain a minimum required dilution of 1:10. The diluted blood sample was centrifuged at 1,100 × g for 10 minutes at RT and the supernatant aliquoted into a clean tube and stored at−80°C until analysis. To calculate plasma concentrations from the diluted whole blood matrix, a dilution factor of 18.18 was applied to correct for the hematocrit component and lysis of red blood cells. Quantitative bioanalysis was performed utilizing a Gyrolab (Gyrolab™ xP, Gyros U.S. Inc.) immunoassay platform. A generic human IgG assay format was utilized, each mAb assay was independently optimized and qualified to give the required sensitivity and dynamic range. mAb standards and controls were prepared in assay buffer. For each assay, inter-day accuracy, precision, selectivity, specificity and dilutional linearity were examined and precision and accuracy of spiked quality controls met pre-established acceptance criteria (%CV ≤20% and %bias ≤30% of nominal). The assay range of quantitation was 23.6 – 5760 ng/mL in 100% mouse plasma for all mAbs tested. The qualification data was generated using Gyrolab Evaluator Software (Version 3.1.5.137).
Quantification of all mAbs in this study used a capture reagent of donkey anti-human IgG (H+L) biotin (Jackson ImmunoResearch Labs; 709–065–149). Bound mAb was detected with Alexa 647-labeled donkey anti-human IgG (H+L) (Jackson ImmunoResearch Labs; 709–605–149). Gyrolab Rexxip F Buffer was used for reagent dilutions (Gyros U.S. Inc.). The concentration of study samples from each mAb were determined by interpolation from a standard curve using a 5-parameter logistic curve fit with 1/y2 response weighting using Watson LIMS Software version 7.4 (Thermo Scientific Inc.). Concentration values below the limit of quantitation were set to 0 ng/mL for all PK calculations.
No potential conflict of interest is reported.
This work was supported by Pfizer.
supplementary_material_1389355.docx
Download MS Word (37.8 KB)Acknowledgments
We would like to express deep gratitude to Dr. Carol Walsh, for her enthusiastic encouragement and help with editing and our very great appreciation to Dr. Yijie Gao, for her useful critiques of this research work. We are also grateful to the following individuals, Richard Zollner, Jocelyn Sanford, Khetemenee Lam, Dave Cirelli, Paul Sakorafas, Jeff Kurz, Minlei Zhang and Eric Bennett for their contribution to this work.
Related Research Data
References
- Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548–58. doi:10.1038/clpt.2008.170. PMID:18784655.
- Acqua WFD, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the Affinity of a Human IgG1 for the Neonatal Fc Receptor: Biological Consequences. The Journal of Immunology. 2002;169:5171–80. doi:10.4049/jimmunol.169.9.5171. PMID:12391234.
- Borrok MJ, Wu Y, Beyaz N, Yu X-Q, Oganesyan V, Dall'Acqua WF, Tsui P. pH-dependent Binding Engineering Reveals an FcRn Affinity Threshold That Governs IgG Recycling. Journal of Biological Chemistry. 2015;290:4282–90. doi:10.1074/jbc.M114.603712. PMID:25538249.
- Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. 2006;281:23514–24. doi:10.1074/jbc.M604292200. PMID:16793771.
- Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. mAbs. 2015;7:483–93. doi:10.1080/19420862.2015.1016696. PMID:25695748.
- Finch DK, Sleeman MA, Moisan J, Ferraro F, Botterell S, Campbell J, Cochrane D, Cruwys S, England E, Lane S, et al. Whole-molecule antibody engineering: generation of a high-affinity anti-IL-6 antibody with extended pharmacokinetics. J Mol Biol. 2011;411:791–807. doi:10.1016/j.jmb.2011.06.031. PMID:21723291.
- Firan M, Bawdon R, Radu C, Ober RJ, Eaken D, Antohe F, Ghetie V, Ward ES. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int Immunol. 2001;13:993–1002. doi:10.1093/intimm/13.8.993. PMID:11470769.
- Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, Ober RJ, Ward ES. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotech. 1997;15:637–40. doi:10.1038/nbt0797-637.
- Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I- related receptor FcRn. Annu Rev Immunol. 2000;18:739–66. doi:10.1146/annurev.immunol.18.1.739. PMID:10837074.
- Kim JK, Tsen MF, Ghetie V, Ward ES. Localization of the site of the murine IgG1 molecule that is involved in binding to the murine intestinal Fc receptor. Eur J Immunol. 1994;24:2429–34. doi:10.1002/eji.1830241025. PMID:7925571.
- Suzuki T, Ishii-Watabe A, Tada M, Kobayashi T, Kanayasu-Toyoda T, Kawanishi T, Yamaguchi T. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: a comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. J Immunol. 2010;184:1968–76. doi:10.4049/jimmunol.0903296. PMID:20083659.
- Ward ES, Zhou J, Ghetie V, Ober RJ. Evidence to support the cellular mechanism involved in serum IgG homeostasis in humans. Int Immunol. 2003;15:187–95. doi:10.1093/intimm/dxg018. PMID:12578848.
- Yeung YA, Wu X, Reyes AE, 2nd, Vernes JM, Lien S, Lowe J, Maia M, Forrest WF, Meng YG, Damico LA, et al. A therapeutic anti-VEGF antibody with increased potency independent of pharmacokinetic half-life. Cancer Res. 2010;70:3269–77. doi:10.1158/0008-5472.CAN-09-4580. PMID:20354184.
- Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28:157–9. doi:10.1038/nbt.1601. PMID:20081867.
- Zhou J, Johnson JE, Ghetie V, Ober RJ, Ward ES. Generation of mutated variants of the human form of the MHC class I-related receptor, FcRn, with increased affinity for mouse immunoglobulin G. J Mol Biol. 2003;332:901–13. doi:10.1016/S0022-2836(03)00952-5. PMID:12972260.
- Jensen PF, Schoch A, Larraillet V, Hilger M, Schlothauer T, Emrich T, Rand KD. A Two-pronged Binding Mechanism of IgG to the Neonatal Fc Receptor Controls Complex Stability and IgG Serum Half-life. Mol Cell Proteomics. 2017;16:451–6. doi:10.1074/mcp.M116.064675.
- Schlothauer T, Rueger P, Stracke JO, Hertenberger H, Fingas F, Kling L, Emrich T, Drabner G, Seeber S, Auer J, et al. Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies. mAbs. 2013;5:576–86. doi:10.4161/mabs.24981. PMID:23765230.
- Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc Natl Acad Sci U S A. 2015;112:5997–6002. doi:10.1073/pnas.1408766112.
- Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos. 2011;39:1469–77. doi:10.1124/dmd.111.039453. PMID:21610128.
- Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93:2645–68. doi:10.1002/jps.20178. PMID:15389672.
- Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature 1994;372:379–83. doi:10.1038/372379a0. PMID:7969498.
- Martin WL, West AP, Jr., Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell. 2001;7:867–77. doi:10.1016/S1097-2765(01)00230-1. PMID:11336709.
- Burmeister WP, Gastinel LN, Simister NE, Blum ML, Bjorkman PJ. Crystal structure at 2.2 A resolution of the MHC-related neonatal Fc receptor. Nature. 1994;372:336–43. doi:10.1038/372336a0. PMID:7969491.
- West AP, Jr., Bjorkman PJ. Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor. Biochemistry. 2000;39:9698–708. doi:10.1021/bi000749m. PMID:10933786.
- Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel. 2010;23:385–92. doi:10.1093/protein/gzq009. PMID:20159773.
- Li B, Tesar D, Boswell CA, Cahaya HS, Wong A, Zhang J, Meng YG, Eigenbrot C, Pantua H, Diao J, et al. Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. mAbs. 2014;6:1255–64. doi:10.4161/mabs.29809. PMID:25517310.
- Myszka DG. Improving biosensor analysis. J Mol Recognit 1999;12:279–84. doi:10.1002/(SICI)1099-1352(199909/10)12:5%3c279::AID-JMR473%3e3.0.CO;2-3. PMID:10556875.
- Rich RL, Myszka DG. Survey of the year. 2007 commercial optical biosensor literature. Journal of Molecular Recognition. 2008;21:355–400. doi:10.1002/jmr.928. PMID:18951413.
- Avery LB, Wang M, Kavosi MS, Joyce A, Kurz JC, Fan Y-Y, Dowty ME, Zhang M, Zhang Y, Cheng A, et al. Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. mAbs. 2016;8:1064–78. doi:10.1080/19420862.2016.1193660. PMID:27232760.
- Gurbaxani B, Dela Cruz LL, Chintalacharuvu K, Morrison SL. Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol Immunol. 2006;43:1462–73. doi:10.1016/j.molimm.2005.07.032. PMID:16139891.
- Thorn M, Piche-Nicholas N, Stedman D, Davenport SW, Zhang N, Collinge M, Bowman CJ. Embryo-Fetal Transfer of Bevacizumab (Avastin) in the Rat Over the Course of Gestation and the Impact of Neonatal Fc Receptor (FcRn) Binding. Birth Defects Research Part B: Developmental and Reproductive Toxicology. 2012;95:363–75. doi:10.1002/bdrb.21026.
- Abdiche YN, Yeung YA, Chaparro-Riggers J, Barman I, Strop P, Chin SM, Pham A, Bolton G, McDonough D, Lindquist K, et al. The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. mAbs. 2015;7:331–43. doi:10.1080/19420862.2015.1008353. PMID:25658443.
- Huber AH, Kelley RF, Gastinel LN, Bjorkman PJ. Crystallization and stoichiometry of binding of a complex between a rat intestinal Fc receptor and Fc. J Mol Biol. 1993;230:1077–83. doi:10.1006/jmbi.1993.1220. PMID:8478919.
- Kim JK, Firan M, Radu CG, Kim CH, Ghetie V, Ward ES. Mapping the site on human IgG for binding of the MHC class I-related receptor, FcRn. Eur J Immunol. 1999;29:2819–25. doi:10.1002/(SICI)1521-4141(199909)29:09%3c2819::AID-IMMU2819%3e3.0.CO;2-6. PMID:10508256.
- Bongini L, Fanelli D, Piazza F, De Los Rios P, Sandin S, Skoglund U. Freezing immunoglobulins to see them move. Proc Natl Acad Sci U S A. 2004;101:6466–71. doi:10.1073/pnas.0400119101. PMID:15082830.
- Bongini L, Fanelli D, Piazza F, De Los Rios P, Sandin S, Skoglund U. Dynamics of antibodies from cryo-electron tomography. Biophys Chem. 2005;115:235–40. doi:10.1016/j.bpc.2004.12.037. PMID:15752611.
- Tian X, Vestergaard B, Thorolfsson M, Yang Z, Rasmussen HB, Langkilde AE. In-depth analysis of subclass-specific conformational preferences of IgG antibodies. IUCrJ. 2015;2:9–18. doi:10.1107/S205225251402209X. PMID:25610623.
- Bumbaca D, Wong A, Drake E, Reyes AE, 2nd, Lin BC, Stephan JP, Desnoyers L, Shen BQ, Dennis MS. Highly specific off-target binding identified and eliminated during the humanization of an antibody against FGF receptor 4. MAbs. 2011;3:376–86. doi:10.4161/mabs.3.4.15786. PMID:21540647.
- Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, White WI, Young JF, Kiener PA. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol. 2007;368:652–65. doi:10.1016/j.jmb.2007.02.024. PMID:17362988.
- Malek A, Sager R, Kuhn P, Nicolaides KH, Schneider H. Evolution of maternofetal transport of immunoglobulins during human pregnancy. Am J Reprod Immunol. 1996;36:248–55. doi:10.1111/j.1600-0897.1996.tb00172.x. PMID:8955500.
- Bloom JW, Madanat MS, Marriott D, Wong T, Chan SY. Intrachain disulfide bond in the core hinge region of human IgG4. Protein Sci. 1997;6:407–15. doi:10.1002/pro.5560060217. PMID:9041643.
- Rayner LE, Hui GK, Gor J, Heenan RK, Dalby PA, Perkins SJ. The Fab Conformations in the Solution Structure of Human Immunoglobulin G4 (IgG4) Restrict Access to Its Fc Region: IMPLICATIONS FOR FUNCTIONAL ACTIVITY. J Biol Chem. 2014;289:20740–56. doi:10.1074/jbc.M114.572404. PMID:24876381.
- Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJR, et al. Complement Is Activated by IgG Hexamers Assembled at the Cell Surface. Science. 2014;343:1260–3. doi:10.1126/science.1248943. PMID:24626930.
- Cook EM, Lindorfer MA, van der Horst H, Oostindie S, Beurskens FJ, Schuurman J, Zent CS, Burack R, Parren PWHI, Taylor RP. Antibodies That Efficiently Form Hexamers upon Antigen Binding Can Induce Complement-Dependent Cytotoxicity under Complement-Limiting Conditions. J Immunol. 2016;197:1762–75. doi:10.4049/jimmunol.1600648. PMID:27474078.
- Hadzhieva M, Pashov AD, Kaveri S, Lacroix-Desmazes S, Mouquet H, Dimitrov JD. Impact of Antigen Density on the Binding Mechanism of IgG Antibodies. Sci Rep. 2017;7:3767. doi:10.1038/s41598-017-03942-z. PMID:28630473.
- Piekarska B, Drozd A, Konieczny L, Król M, Jurkowski W, Roterman I, Spólnik P, Stopa B, Rybarska J. The Indirect Generation of Long-distance Structural Changes in Antibodies upon their Binding to Antigen. Chem Biol Drug Des. 2006;68:276–83. doi:10.1111/j.1747-0285.2006.00448.x.
- Jensen PF, Larraillet V, Schlothauer T, Kettenberger H, Hilger M, Rand KD. Investigating the Interaction between the Neonatal Fc Receptor and Monoclonal Antibody Variants by Hydrogen/Deuterium Exchange Mass Spectrometry. Mol Cell Proteomics. 2015;14:148–61. doi:10.1074/mcp.M114.042044.
- Gurbaxani B, Dostalek M, Gardner I. Are endosomal trafficking parameters better targets for improving mAb pharmacokinetics than FcRn binding affinity? Mol Immunol. 2013;56:660–74. doi:10.1016/j.molimm.2013.05.008. PMID:23917469.
- Unverdorben F, Richter F, Hutt M, Seifert O, Malinge P, Fischer N, Kontermann RE. Pharmacokinetic properties of IgG and various Fc fusion proteins in mice. mAbs. 2016;8:120–8. doi:10.1080/19420862.2015.1113360. PMID:26514880.
- Joyce AP, Wang M, Lawrence-Henderson R, Filliettaz C, Leung SS, Xu X, O'Hara DM. One Mouse, One Pharmacokinetic Profile: Quantitative Whole Blood Serial Sampling for Biotherapeutics. Pharm Res. 2014;31:1823–33. doi:10.1007/s11095-013-1286-y. PMID:24464271.