
ABSTRACT
Agonism of members of the tumor necrosis factor receptor superfamily (TNFRSF) with monoclonal antibodies is of high therapeutic interest due to their role in immune regulation and cell proliferation. A major hurdle for pharmacologic activation of this receptor class is the requirement for high-order clustering, a mechanism that imposes a reliance in vivo on Fc receptor-mediated crosslinking. This extrinsic dependence represents a potential limitation of virtually the entire pipeline of agonist TNFRSF antibody drugs, of which none have thus far been approved or reached late-stage clinical trials. We show that tetravalent biepitopic targeting enables robust intrinsic antibody agonism for two members of this family, OX40 and DR5, that is superior to extrinsically crosslinked native parental antibodies. Tetravalent biepitopic anti-OX40 engagement co-stimulated OX40low cells, obviated the requirement for CD28 co-signal for T cell activation, and enabled superior pharmacodynamic activity relative to native IgG in a murine vaccination model. This work establishes a proof of concept for an engineering approach that addresses a major gap for the therapeutic activation of this important receptor class.
Introduction
Receptors of the tumor necrosis factor receptor superfamily (TNFRSF) are central regulators of immunity, cell proliferation and death, and other important biological processes. Because of these essential roles, targeted agonism of this receptor class is an active therapeutic approach for the treatment of cancer, autoimmunity, and other diseases.Citation1–Citation4 Because cognate ligands of these receptors generally make suboptimal drugs due to poor production, stability, or pharmacokinetics (PK), the monoclonal antibody has been the most common drug modality advanced for this receptor class. A significant challenge for the pharmacologic agonism of these receptors by antibodies is the general requirement for high-order receptor clustering,Citation5,Citation6 whereby a multiplicity of receptors are brought together in close proximity on the cell surface. Antibodies achieve this mechanism in pre-clinical in vivo models via extrinsic cross-linking from Fc engagement with Fc gamma receptors (FcγRs) on immune cells.Citation7–Citation9 Despite the advancement of a large pipeline of TNFRSF agonist antibodies into development, as yet none have been approved or reached late-stage clinical trials, and several have been withdrawn.Citation1,Citation3,Citation4,Citation10–Citation12 While a diverse host of factors may be responsible, the lack of intrinsic potency represents a major deficiency of this class of agents, and it remains a question as to whether a native antibody is capable of sufficiently driving receptor signaling in a therapeutic setting.
Here, we describe a simple yet effective engineering solution to this Achilles’ heel of agonist antibodies. Using a diverse series of engineered antibody formats, we demonstrate consistently that optimal TNFRSF agonist activity is enabled by engaging two distinct epitopes in the context of a tetravalent molecule. With both cell lines and primary human cells, we demonstrate that these tetravalent biepitopic antibodies mediate robust agonism activity that does not rely on extrinsic crosslinking. Using biophysical methods, microscopy, and crystallographic studies, we provide insights into the clustering mechanism of this approach. Finally, we validate in a relevant in vivo model that this engineering strategy enables pharmacodynamic (PD) activity that is both enhanced relative to a native IgG1 and independent of FcγR-mediated crosslinking.
Results
Engineering of multivalent and multiepitopic antibody formats
Antibodies are naturally capable of promoting immune complexes through a combination of multivalency and polyclonality. We hypothesized that a combination of multivalent and multiepitopic engagement of TNFRSF receptors could provide the necessary clustering to drive TNFRSF signaling. We chose the TNFRSF members OX40 (TNFRSF4/CD134) and Death Receptor 5 (DR5/TNFRSF10B), two targets of therapeutic interest for which agonist antibodies are dependent on FcγR-mediated crosslinking,Citation9,Citation13 but which have not realized clinical success.Citation3,Citation10 Recombinant engineering and bioconjugation approaches were used to develop a set of four tetravalent biepitopic and monoepitopic antibody formats (). Recombinant (r:) versions comprised either antigen-binding fragment (Fab) regions (r:Fab-IgG)Citation14 or variable fragment (Fv) regions (r:Fv-IgG)Citation15 linked genetically to an IgG via a flexible amino acid linker. Conjugated (c:) versions were generated by chemically coupling either two Fabs to an IgG (c:Fab-IgG) or two IgGs to each other (c:IgG-IgG) through engineered free cysteines using a bis-maleimide linker. Mono- and biepitopic versions of all four formats targeting OX40 were generated using two humanized antibodies, Ab1 and Ab2, that bind distinct epitopes on OX40 at 0.45 nM and 2.5 nM, respectively. These two antibodies were selected solely because they bound separate epitopes, and no screening was done a priori for their ability to synergize in combination. We also produced a hexameric version of these antibodies using an Fc variant E345R/E430G/S440Y (referred to as RGY) previously described.Citation16,Citation17 Native IgG1 (Ab1 and Ab2) and bivalent biepitopic (Ab1/Ab2) antibodies () were produced as controls, and select versions were produced with Fc substitutions L234A/L235A/P329G (LALAPG)Citation18 to ablate binding to all FcγRs. Solution and cell surface affinities are provided in Supplementary Table 1. Production of the antibody formats ranged in complexity. All antibodies and antibody formats were purified to greater than 99% monomeric species (see Materials and Methods), and remained monodisperse in solution after purification.
Figure 1. Illustration of antibody formats in the present study. Yellow represents the variable region of Ab1, orange represents the variable region of Ab2, and blue represents constant regions. “r:” represents recombinantly linked formats, and “c:” represents chemically coupled formats. r:Fv-IgG variable domains were linked recombinantly using a 10-residue Gly-Ser linker (GGGGSGGGGS), r:Fab-IgG Fabs were linked recombinantly using a 7 residue Gly-Ser linker (GGGGSGG), and coupled formats c:Fab-IgG and c:IgG-IgG were linked with thiol chemistry through a bis-maleimido polyethylene glycol (BMPEG) linker containing 3 PEG units. The labels above the formats provide arbitrary designations for the monoepitopic (left hand side) and biepitopic (right-hand side) versions of each format. The bracketed formats were not characterized in the present study but are included in the illustration for completeness.
Tetravalent biepitopic antibodies promote receptor signaling without relying on extrinsic crosslinking
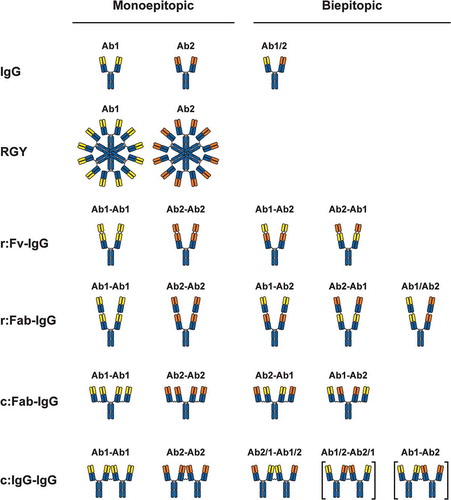
Anti-OX40 antibodies were tested for their ability to promote receptor signaling using engineered Jurkat cells expressing human OX40 and an NF-κB luciferase reporter (–), EC50 values are provided in Supplementary Table 1). In the absence of extrinsic crosslinking, no agonism was mediated by the parental antibodies or the bivalent biepitopic Ab1/Ab2 antibody ()). Crosslinking either antibodies or ligand with a secondary antibody resulted in potent receptor activation, with substantially reduced activity at higher concentrations. Increasing valency to 12 using the noncovalent Fc variant RGY resulted in intrinsic activation, consistent with previous work.Citation16,Citation17,Citation19 While tetravalent monoepitopic targeting (red diamonds) provided a modest level of agonism independent of extrinsic crosslinking, tetravalent biepitopic targeting (blue circles) provided a more profound level of intrinsic activity (–)). The same trend in tetravalent biepitopic superiority was observed across the four distinct antibody formats, and either Fv orientation of the r:Fv-IgG and r:Fab-IgG formats demonstrated comparable enhanced activity (,)). The two biepitopic recombinant formats (r:Fv-IgG and r:Fab-IgG) were slightly higher in activity than the two chemically coupled formats (c:Fab-IgG and c:IgG-IgG). In contrast to extrinsically crosslinked ligand or IgG, the intrinsic agonist formats displayed only modest or no reduction in activity at higher concentrations.
Figure 2. Tetravalent biepitopic antibody formats enable intrinsic agonism of TNFRSFs. (a) NF-κB signaling mediated by OX40L-huIgG1 Fc fusion (OX40L) and anti-OX40 antibody formats. IgG1 (squares) and OX40L (downward triangles) were tested with (+XL) and without extrinsic secondary crosslinking. Orange triangle represents a bivalent biepitopic antibody (Ab1/Ab2), and upward triangles represent hexameric variant antibodies (RGY). (b–e) OX40 NF-κB signaling activity mediated by tetravalent biepitopic (blue) and monoepitopic (red) antibody formats: r:Fv-IgG (b), r:Fab-IgG (c), c:Fab-IgG (d) or c:IgG-IgG (e). (f) DR5 driven caspase-8 activity and (g) anti-proliferative activity in COLO 205 cells mediated by multivalent and multiepitopic antibody formats and soluble Flag-Apo2L. For all tetravalent formats (b–g) blue circles indicate biepitopic formats and red diamonds indicate monoepitopic formats, respectively. The x-axis for each graph represents the molar concentration of each complete molecule irrespective of number of receptor binding units; i.e., 1 nM of a bivalent antibody has two binding sites, whereas a 1 nM equivalent of an r:Fv-IgG has four binding sites.
To explore the broader potential of the tetravalent biepitopic targeting approach, format antibodies to another TNFRSF member death receptor 5 (DR5) were generated and tested. Building on our results with the anti-OX40 biepitopic formats, we explored a panel of 14 antibodies across 3 epitope bins for pairs that intrinsically agonized DR5 in the r:Fab-IgG format. We selected a lead r:Fab-IgG comprising two antibodies (Ab1 and Ab2) that bind distinct epitopes on the receptor at 0.89 and 0.32 nM, respectively. Native and engineered anti-DR5 antibodies were tested for their ability to elicit caspase-8 signaling ()) and anti-proliferative activity ()) against a COLO 205 colon carcinoma cell line. The EC50 values for the curves and DR5 binding data are provided in Supplementary Table 2. Consistent with the anti-OX40 results, tetravalent biepitopic targeting provided intrinsic apoptotic signaling and anti-proliferative activity, in contrast to parental and bivalent biepitopic IgGs that had no or weak activity in the absence of crosslinking. The tetravalent biepitopic r:Fab-IgG was superior in both potency and maximal activity to the tetravalent monoepitopic Ab1-Ab1 version, which showed some intrinsic DR5 agonism; the tetravalent monoepitopic Ab2-Ab2 version was completely inactive in the absence of crosslinking. Together, these results suggest that the application of tetravalent biepitopic may extend to other TNFRSF members.
Tetravalent biepitopic anti-OX40 antibodies co-stimulate human T cells in the absence of extrinsic crosslinking and CD28 co-signal
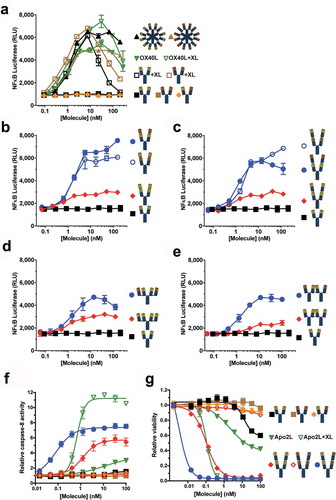
The relative activities of the anti-OX40 antibodies were tested for their ability to activate primary human CD4+ T cells (). OX40 co-stimulation requires both T cell receptor (TCR) engagement with peptide-loaded major histocompatibility complex (MHC) and CD28 co-signal provided by B7-1 (CD80) or B7-2 (CD86) on antigen-presenting cells (APCs).Citation20 In our assay, an anti-CD3 antibody provided TCR signal, and co-cultured L cells expressing human B7-1 and FcγRIIa (CD32a) or FcγRIIa alone were used as surrogate APCs. In the presence of FcγR-mediated crosslinking, native IgG1 Ab1 mediated a moderate level of T cell co-stimulation in the presence (B7-1+, ,)) but not absence (B7-1−, ,)) of CD28 co-signal. In contrast, biepitopic r:Fv-IgG promoted robust T cell activation in the presence or absence of B7-1. This result was due at least in part to the ability of the tetravalent biepitopic format to co-activate OX40low T cells ()). Consistent with the NF-κB reporter data (), both r:Fv-IgG orientations (Ab1-Ab2 and Ab2-Ab1) provided comparable levels of activity (,)). Removal of Fc-mediated crosslinking using the LALAPG variant (,)) demonstrated the intrinsic activity of the format and the comparable level of activity mediated by tetravalent biepitopic targeting independent of whether extrinsic crosslinking or CD28 co-signal is present.
Figure 3. Tetravalent biepitopic targeting enables T cell activation without extrinsic crosslinking and obviates requirement for CD28 co-signal. (a–d) Co-stimulation of CD3-activated primary human CD4+ T cells by anti-OX40 IgG1 (a and c) and Fc attenuated LALAPG (b and d) versions of r:Fv-IgG in the presence of co-cells expressing FcγRIIa and B7-1 (FcγRIIa+ B7-1+) (A and B) or FcγRIIa alone (FcγRIIa+ B7-1−) (c and d). (e) Provision of CD28 signal (B7-1+) augments OX40 expression on T cells. Primary human CD4+ memory T cells were co-cultured with B7-1− (black) or B7-1+ (red) L cells over time. (f and g) Co-stimulation of primary human CD4+ T cells by hexameric anti-OX40 antibodies in the presence of FcγRIIa+ B7-1+ (f) or FcγRIIa+ B7-1− co-cells (g). Closed symbols represent IgG1 versions and open symbols represent Fc attenuated LALAPG versions for both IgG1 (black) and RGY (brown) versions. (h–j) Co-stimulation of primary human CD4+ T cells by IgG1 versions of tetravalent monoepitopic and biepitopic formats r:Fab-IgG (h), c:Fab-IgG (i), and c:IgG1-IgG (j) in the presence of FcγRIIa+ B7-1− co-cells. For data on all tetravalent formats (a–d, h–j), blue circles indicate biepitopic formats and red diamonds indicate monoepitopic formats, respectively, black squares indicate Ab1 IgG1, and brown triangles indicate Ab1 IgG1 RGY comparators. The x-axis for each graph represents the molar concentration of each complete molecule irrespective of number of receptor binding units; i.e., 1 nM of a bivalent antibody has two binding sites, whereas a 1 nM equivalent of an r:Fv-IgG has four binding sites. Y-axes are scaled the same for samples run in the same internally controlled experiment (same blood donor, same 7-day culture) as follows: (a–d) were executed in the same experiment, (f–g) were executed in the same experiment, and (h–j) were executed in the same experiment. RGY IgG1 (solid gold triangles) was used as a standard for comparison of results across different experiments.
In contrast, whereas monoepitopic versions of the r:Fv-IgG (red diamonds) mediated strong activity in the presence of Fc-mediated crosslinking and CD28 co-signal ()), activity in the absence of B7-1 was only modest ()) and virtually dependent on FcγR-mediated crosslinking (LALAPG versions, ,)). Mixtures of the parental IgG1 versions of Ab1 and Ab2 (1:1 pre-mixed IgGs over a concentration range) did not enable intrinsic agonism, and no additive activity beyond the parental antibodies was detected when the 1:1 mixture was crosslinked (data not shown), indicating that covalent linkage of the two variable regions is required for the tetravalent formats. Similar to biepitopic r:Fv-IgG format, hexameric targeting of OX40 using the RGY variant Ab1 demonstrated enhancement of activity relative to IgG1 that was both independent of FcγR-mediated crosslinking and CD28 co-signal (,)). A trend of slightly weaker EC50 values was observed for r:Fv-IgG and RGY hexamer formats in B7-1+ vs. B7-1− assays, respectively (Supplementary Table 1), possibly reflecting the lower OX40 expression levels on T cells in the absence of CD28 co-stimulation. In the absence of anti-CD3, neither the r:Fv-IgG nor the hexamer format promoted T cell proliferation (Supplementary Figure 1). Dependence on TCR-signal is a key safety element of T cell co-stimulatory drugs.Citation21,Citation22 Experiments with the r:Fab-IgG ()), c:Fab-IgG ()), and c:IgG-IgG ()) formats in the FcγRIIa+ B7-1− co-stimulation assay agreed well with the r:Fv-IgG results, consistently demonstrating robust T cell activation from the tetravalent biepitopic, but not monoepitopic formats and non-reliance on provision of CD28 co-signal. Expanded experiments with the r:Fab-IgG (Supplementary Figure 1) showed similar results to the r:Fv-IgG, demonstrating more broadly the independence of tetravalent biepitopic activity on extrinsic crosslinking and CD28 co-signal, as well as the comparability of the two orientations.
Tetravalent biepitopic targeting clusters antibodies and receptors into high-order complexes
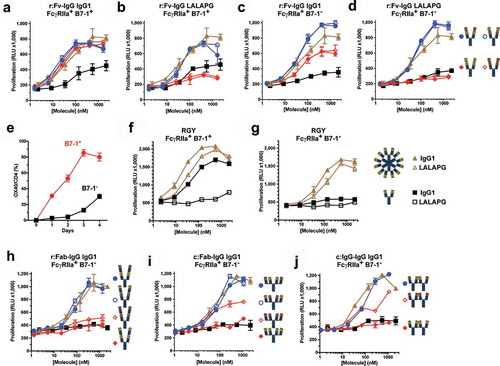
To investigate the mechanism of intrinsic agonism, the ability of the antibodies to form high-order immune complexes upon binding to purified OX40 was tested using size exclusion chromatography (SEC). A shift to earlier elution time was observed for complexes formed between biepitopic relative to monoepitopic r:Fv-IgG ()). Similar chromatographic results were obtained for r:Fab-IgG and c:Fab-IgG formats (data not shown). The size of the largest antibody/OX40 complexes was measured using SEC linked in-line with multiple angle light scattering (SEC-MALS). The data ()) further supported the larger immune complex formation by tetravalent biepitopic relative to monoepitopic engagement, and indicated that the r:Fv-IgG format promoted the largest complexes. The importance of both tetravalency and biepitopic binding was illustrated further by the almost negligible shift in complex size observed from the bivalent biepitopic IgG1 (Ab1/Ab2, right-most blue bar in )), consistent with the inability of the bivalent biepitopic Ab1/Ab2 format to intrinsically agonize OX40 or DR5 in the cell-based assays (,,)).
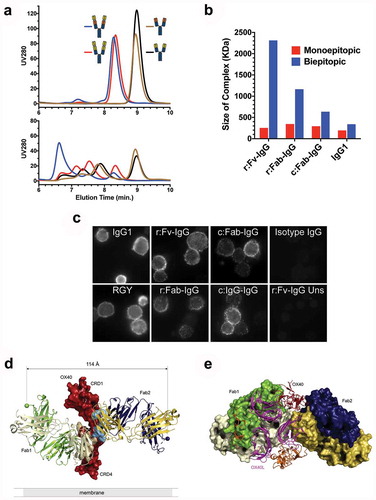
Figure 4. Tetravalent biepitopic targeting promotes higher order immune complexes. (a) SEC chromatograms for native IgG1 Ab1 (black) and Ab2 (brown), r:Fv-IgG biepitopic (blue), and r:Fv-IgG monoepitopic (red) antibodies in the absence (top graph) or presence (bottom graph) of OX40 ECD protein. (b) Size of the largest antibody/OX40 complex calculated by SEC-MALS. Biepitopic IgG1 (right-most blue bar) represents the bivalent biepitopic IgG Ab1/Ab2 illustrated in . (c) Immunofluorescence microscopy of anti-CD3 stimulated primary T cells following binding of 6.7 nM anti-OX40 antibodies on ice, fixation, and detection with Cy3-conjugated anti-human antibody. The assay was conducted on ice to prevent internalization or degradation, and thus the data reflect only differential binding and clustering of the antibody formats on the cell surface. Binding specificity was evidenced by lack of signal on stimulated T cells with an isotype IgG control (upper right), and by r:Fv-IgG on unstimulated (OX40-negative) T cells (lower right). IgG1 (upper left) and RGY (lower left) represent Ab1 IgG1 and Ab1 RGY antibodies, respectively. (d and e) Crystallographic structure of ternary complex of OX40 bound by Ab1 and Ab2 Fabs. (d) Crystallographic structure of Ab1 (green/ivory) and Ab2 (navy/gold) Fabs bound to OX40 (red). Spheres mark the C-termini of the heavy chain components where attachment to the hinge region would occur in the context of a full-length IgG, and the distance (114 Å) between those two C-termini is labeled. Epitopes are shaded on the molecular surface in tan (Ab1) and light blue (Ab2). For the presented OX40 orientation the cell membrane would be at the lower side of the image. (e) “Top down” view of the ternary complex with Ab1 and Ab2 Fabs rendered in molecular surface bound to the OX40 ECD in red. The OX40:OX40L trimer (OX40 in orange, OX40L in magenta) derived from PDB 2HEV has been superposed by alignment of the OX40 chain (the aligned 2HEV OX40 is not shown for clarity), indicating extensive steric clash between either of the Fabs and the physiological trimeric OX40L arrangement.
Cross-linking of OX40 by the tetravalent biepitopic formats could also be visualized on the cell surface. When activated T cells were incubated with antibodies, apparent clustering was readily evident with the four biepitopic formats and to a lesser extent monoepitopic RGY, in contrast to the smooth surface signal seen with native IgG1 ()).
Structure of ternary Fab/Fab/receptor complex reveals epitope insights into clustering mechanism
To further understand the mechanism by which tetravalent biepitopic targeting promotes intrinsic receptor agonism, we determined the crystallographic structure of the ternary complex of OX40 extracellular domain bound to Ab1 and Ab2 Fabs at 2.6 Å resolution (,e), ). The two antibodies bind OX40 receptor on diametrically opposed regions, with Ab1 Fab (referred to as Fab1) binding to cysteine-rich domain (CRD) 3 of OX40 between residues 114 to 142 and the Ab2 Fab (referred to as Fab2) binding to CRD 2 between residues 68 to 98 ()). The calculated distance between the heavy chain C-termini of the two Fabs in the structure is ~114 Å. In the context of a full-length IgG1, using the most N-terminal hinge disulfide at EU position Cys226 as the tether point and a random coil residue distance of 3.5 Å, the C-termini of the two IgG Fab arms are constrained to an approximate maximal distance of ~35 Å relative to each other. This analysis, as well as the inverted rotational orientation of the two Fabs relative to each other, suggests that the orientation of the two Fabs does not permit intra-IgG OX40 binding, forcing inter-receptor interactions that would ostensibly promote high-order complexation.
Table 1. Data collection and refinement statistics for OX40:Fab binary and ternary complexes.
In addition to the ternary complex, we also solved the structures of the binary OX40:Fab1 and OX40:Fab2 complexes at 3.25 and 2.1 Å resolution, respectively (Supplementary Figure 2, ). The overall OX40 conformation and the disposition of the Fabs relative to OX40 in the binary complexes are similar to those in the ternary complex, save for a ~ 30º shift in the elbow angle of Ab2 Fab between the variable and constant regions (Supplementary Figure 3). This deviation of the CH1 and CL region may be attributed to crystal packing differences. In the binary complexes, the OX40 receptor lacked order distal to the Fab binding interfaces, and consequently it was not possible to model all CRDs (Supplementary Figure 2). In contrast, the OX40 receptor is more ordered in the ternary complex and all four CRDs are present in the structure.
The global conformation of OX40 receptor in the ternary OX40:Fab1:Fab2 complex is consistent with that in the OX40:OX40L complex structure (PDB 2HEVCitation23), which also has order for all four CRDs (Supplementary Figure 2(c,d)). The exception is a somewhat malleable amino-terminal portion of CRD1 and a notable movement of an exposed loop on CRD3. This loop from Pro115 to Pro121 includes Tyr119, which pivots ~10Å from its OX40L-interacting position (predominantly with Asn166 and Gly167) to interact with the Ab1 Fab at Glu1 and Gly26, a location that would be obstructed by a neighboring OX40L in the trimeric arrangement (Supplementary Figure 4). Aside from these finer contact details, comparison between the OX40:OX40 and ternary OX40:Fab1:Fab2 structures indicates that the Ab1 and Ab2 epitopes overlap the OX40 interaction surfaces of the physiological ligand OX40L ()), suggesting that Fab-mediated agonism may proceed by distinct arrangements of the OX40 receptor relative to the OX40L trimer.
Engineered antibody formats comprising a single Fc region display normal PK in mice
The PK of the antibody formats was assessed in immune-competent C57BL/6 mice ()). The three tetravalent formats containing a single IgG1 Fc region (r:Fv-IgG, r:Fab-IgG, c:Fab-IgG) showed PK profiles comparable to native IgG1 control. In contrast, the c:IgG-IgG that contains two Fc domains showed PK comparable to native IgG1 only up to day 3 post-dose, but a rapid drop in serum concentrations thereafter. The delayed drop in serum exposure in C57BL/6 mice and uncompromised exposure for c:IgG-IgG in SCID mice (Supplementary Figure 5) suggests that the drop in exposure 3 days post-dose is caused by anti-drug antibody response.Citation24 All formats remained intact in vivo based on mass spectrometry data of serum samples (data not shown). In contrast to the tetravalent formats, the RGY hexamer showed rapid clearance in both immune competent ()) and SCID mice (Supplementary Figure 5). Other work has demonstrated that different Fc-engineered variants that do not form solution hexamers can retain intrinsic agonist activity, yet possess more normal clearance properties in SCID mice.Citation19 While we have generated similar results with a broader panel of Fc-engineered variants that behave as monomeric or hexameric antibody species, we have found the in vivo activity and disposition properties of Fc-engineered variants that promote Fc hexamerization to be complex (data not shown), and have therefore limited the scope of the current report. Overall, while further investigation is needed to understand the basis of these results, together our data suggest that multiple Fc regions can present PK or immunogenicity liabilities for multivalent antibody formats.
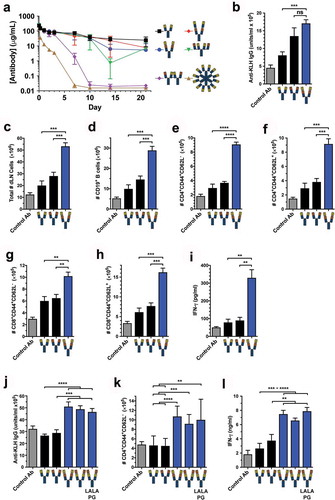
Figure 5. Tetravalent biepitopic OX40 targeting enables superior in vivo pharmacodynamic activity. (a) PK of native IgG1, tetravalent formats, and RGY hexamer in C57BL/6 mice at 10 mg/kg i.v. dose. Data are mean and SD, n = 3/time point/group. (b–e) Pharmacodynamic activity of native IgG1 and biepitopic r:Fv-IgG (Ab1-Ab2 orientation) antibodies in response to KLH-immunization in hOX40ki mice. Control Ab represents immunization with CFA/KLH and anti-gD human IgG1. (b) Anti-KLH IgG in serum on day 14 post immunization measured by ELISA. (C-H) Flow cytometry was used to determine on day 20 total draining lymph node (dLN) cells (c), CD19+ B cells (d), effector memory (CD44+ CD62L−) CD4+ T cells (e), central memory (CD44+ CD62L+) CD4+ T cells (f), effector memory CD8+ T cells (g), and central memory CD8+ T cells (h). (i) KLH-specific recall response was measured by in vitro stimulation of dLN cells with KLH; interferon gamma (IFN-γ) production was assessed in the supernatant after 5 days by luminex. (j–l) In a separate in vivo experiment, an expanded set of r:Fv-IgG antibodies was tested that included both Fv orientations (Ab1-Ab2 and Ab2-Ab1) and the Fc attenuated LALAPG version of the Ab2-Ab1 r:Fv-IgG. (J) Anti-KLH IgG in serum on day 14 post immunization measured by ELISA. (k) Number of effector memory CD4+ T cells on day 20. (l) IFN-γ production from ex vivo KLH-stimulated dLN cells. ****P < .0001; ***P < .001; **P < .01; ns = not significant (P > .05); Two-tailed P values from unpaired t-test. For PD experiments all data are mean and standard error of the mean (SEM); for the experiment presented in (b–i) n = 3 for Control Ab and n = 5 for all other antibody test articles; for the experiment presented in (j–l) n = 8 for all test articles.
Tetravalent biepitopic anti-OX40 antibodies promote enhanced T cell response in vivo that is non-reliant on Fc interactions
The anti-OX40 r:Fv-IgG (Ab1-Ab2 orientation) was tested for PD activity in human OX40-knockin (hOX40ki) mice. These mice showed hOX40 expression on CD4+, but not CD8+ T cells (Supplementary Figure 6). Accordingly, we tested the ability of the engineered antibody to stimulate CD4+ T cell responses to immunization with keyhole limpet hemocyanin (KLH). Relative to the Ab1 IgG1 control, the tetravalent biepitopic format showed enhanced anti-KLH IgG response ()). To access memory responses, mice were intraperitoneally rechallenged on Day 16 with soluble KLH, and draining lymph nodes were profiled 4 days post-rechallenge (Day 20). Relative to both IgG1 parental antibodies, the tetravalent biepitopic format showed superior expansion of total cells ()), B cells ()), effector (CD44+ CD62L−) and central memory (CD44+CD62L+) CD4+ T cells (,f)), and effector and central memory CD8+ T cells (,h)). The tetravalent biepitopic-treated group also showed greater recall responses as measured by interferon gamma (IFN-γ) after ex vivo restimulation of draining lymph node (LN) cells with KLH ()). Expanding the analysis to include a broader r:Fv-IgG set (–l)) demonstrated superior costimulatory activity of both Fv orientations (Ab1-Ab2 and Ab2-Ab1) over the parental IgGs, and maintenance of enhanced activity in the LALAPG version of the format, thus supporting the independence of activity on Fc-mediated interactions.
Discussion
Monoclonal antibodies are used as receptor agonists for research and therapeutic applications generally to mimic the cell signaling properties of a natural ligand.Citation25 As drugs, antibodies can have advantages over natural ligands, including high production, good stability, favorable PK properties, and extensive manufacturing and clinical development experience.Citation26 In contrast to conventional receptor–ligand antagonism, which plays to the strength of antibodies and generally follows more classical pharmacology, agonism can require more complex mechanistic elements depending on the nature of the receptor biochemistry. For some receptors, activation is promoted by ligand-induced dimerization, a mechanism that in principle can be readily mimicked by a bivalent IgG. In contrast, some receptors require higher-order crosslinking to signal, such as when cognate ligands are multimeric or where clustering of ligand-receptor complexes is driven by cell–cell interactions, as occurs for example within the immune synapse at the interface between T cells and antigen-presenting cells. Members of the TNFRSF are perhaps the most widely studied of this mechanistic class and are common antibody targets for drug development owing to their important roles in regulating cellular activation and proliferation.
Whereas crosslinking of agonist anti-TNFRSF antibodies can be achieved in vitro by artificially coating on plates or using secondary reagents, in vivo crosslinking and activity generally rely on Fc engagement with FcγRs on immune cells. While FcγR-mediated crosslinking may be plentiful in pre-clinical models, there is little clinical evidence that agonist antibodies broadly achieve sufficient Fc-mediated crosslinking at their site of action. Complicating the picture further is the potential in some cases for engagement of both FcγR and target receptor on the same cell surface (cis co-engagement, e.g., CD40, 4-1BB, or CD27 on B cells, monocytes, or natural killer cells), versus the absence of FcγRs on the target cell, and thus requirement for antibody-mediated cell–cell interactions (trans co-engagement, e.g., OX40, GITR, 4-1BB, or CD27 on T cells). Overall, despite intense clinical development of agents attempting to activate TNFRSFs for therapy,Citation3,Citation4,Citation27 the broader pipeline of anti-TNFRSF agonist antibodies has thus far not demonstrated success, and as yet none have been approved or reached pivotal trials. While the biology of each individual target plays a central role in success or failure, all of these agents suffer from the same pharmacologic hurdle, namely that their activity relies on extrinsic FcγR-mediated crosslinking by resident immune cells.
Our study advances an antibody engineering solution for the pharmacologic agonism of this important therapeutic target class. Our approach is inspired by and leverages a mechanism of immunoglobulins accessed by natural biology through multivalency and polyclonality. We show that tetravalent biepitopic targeting provides robust intrinsic agonist activity in vitro and in vivo, in an antibody format with IgG-like solution properties. The SEC-MALS and microscopy data indicate that the tetravalent biepitopic antibody formats promote high order receptor complex formation both in solution and on the cell surface. Structural analysis provides insight as to why the two particular antibodies tested may disfavor intra-OX40 engagement to promote inter-OX40 complexes. Intuitively, monoepitopic target engagement terminates antibody/receptor complexes at a stoichiometry equal to the valency. In contrast, biepitopic engagement is theoretically able to promote complexes that propagate, the two binding sites enabling daisy-chained interactions as long as intra-receptor binding by antibody arms is disfavored. In effect, the tetravalent biepitopic approach described here enables an immune complex mechanism like that which occurs naturally via IgG polyclonality, but with a monoclonal approach that is amenable to biotherapeutic development. Multiepitopic targeting and other engineered mechanisms that enhance receptor clustering have applications beyond agonists. For example, previous work successfully demonstrated that biepitopic engagement of human epidermal growth factor receptor 2 can drive increased receptor internalization to enhance the anti-tumor activity of an antibody drug conjugate.Citation28 In our study, the superior activity consistently observed for two TNFRSFs and across four distinct tetravalent formats suggests that the mechanism is robust. Work in progress that explores engineering parameters more deeply suggests that intrinsic agonism and activity enhancement are achievable across a spectrum of flexibility, geometry, and epitope.
A surprising result of our engineering approach is the capacity to drive agonism of OX40low T cells in a manner that does not require CD28 signal. OX40, like many receptors in the TNFRSF class, is generally characterized as a third stimulatory signal (signal 1 = TCR, signal 2 = CD28, signal 3 = OX40 or other co-stimulatory receptor). Upon TCR engagement, CD28-mediated co-stimulation is required to prevent a refractory anergic state in T cells.Citation29 Productive T cell activation in the absence of CD28 stimulation has been reported in autoimmunity where pathogenic T cells express high-affinity TCR, augmenting signal 1 and bypassing the need for co-stimulation.Citation30 Accordingly, therapeutic approaches have focused on blocking co-stimulatory interactions for autoimmunity, not agonizing them for oncology. To our knowledge, there is no published literature on activation of OX40 that obviates CD28 co-signal, or any demonstration that with appropriate stimulation this signal 3 can effectively be converted to signal 2. The potential of such CD28-independent activity could be most valuable for the treatment of weakly inflamed tumorsCitation31 where expression of co-stimulatory receptors and ligands may be low. Indeed, recent studies demonstrate a central role of CD28 co-signal in the mechanism of action of PD-1-targeted therapies.Citation32,Citation33 A potential issue related to such activity is safety. Lowering the bar for activation increases the risk for toxicity, including potentially organ-specific autoimmunity, as has been observed with other immunomodulatory approaches such as CTLA-4 blockade.Citation34 Regardless, the ability of tetravalent biepitopic targeting to drive agonism of OX40low T cells in a manner that obviates CD28 signal illustrates the potential of engineered agonist formats to access high-bar pharmacological activities beyond native IgGs.
While this work establishes a general principle and shows proof of concept, therapeutic validation requires further study. More broadly, the advancement of engineered approaches to antibody receptor agonism is timely. That there are sufficient FcγRs at sites of action to enable clinical activity is a critical assumption of virtually the entire pipeline of anti-TNFRSF agonist antibodies. As a consequence, the current generation of anti-TNFRSF agonists is not optimally designed to conclusively test the biological and clinical hypotheses of activating any one individual target receptor. Independent of the particular antibody platform approach, our hope is that this work will influence progression from native Fc-reliant IgGs towards optimally engineered formats with intrinsic activity for agonizing receptors of therapeutic potential.
Materials and methods
Construction and production of antibodies
Variable regions of anti-OX40 Ab1 and Ab2, anti-DR5 Ab1 and Ab2, and the anti-gD (glycoprotein D of Herpes Simplex Virus) negative control were derived in-house. Molecular biology was carried out using either gene synthesis (Genewiz) or through mutagenesis using QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Cat. 210514) or Q5 Site-Directed Mutagenesis Kit (New England BioLab Cat. E0554S). DNA encoding antibody heavy and light chains were constructed in the pRK mammalian expression vector.Citation35 Heavy chains were constructed with human IgG1 constant chains that were native or contained Fc variants L234A/L235A/P329G (LALAPG) for Fc effector function attenuation,Citation18 E345R/E430G/S440Y (RGY) for hexamerization,Citation16 or either T366W (knob) or T366S/L368A/Y407V (hole) for Fc heterodimerizationCitation36 (EU numbering). Additional variants used in the context of different antibody formats are provided in the respective sections below.
Native IgG1 and r:Fv-IgG antibodies
Monoepitopic and biepitopic recombinant Fv-IgG (r:Fv-IgG) anti-OX40 antibodies were constructed by genetically engineering an additional VL region N-terminal to the native VL region, and an additional VH region N-terminal to the native VH region.Citation15 Variable domains were connected using a 10-residue Gly-Ser linker (GGGGSGGGGS). Irrespective of whether linked variable regions were monoepitopic (Ab1-Ab1 and Ab2-Ab2) or biepitopic (Ab1-Ab2 and Ab2-Ab1), all r:Fv-IgGs are similar to native IgGs in that they are a “2 DNA system”, namely production was accomplished by co-transfecting DNAs for 1 heavy chain and 1 light chain. Consequently, no additional engineering was needed to control heavy and light chain pairing.
For all IgG1 and r:Fv-IgG antibodies, the heavy and light chain DNAs in pRK vector were cotransfected into HEK293 or Chinese hamster ovary (CHO) cells for expression. Harvested media was loaded onto a 5 mL MabSelect SuRe column (GE Healthcare, #17–5438-01). The loaded column was washed with 10 column volumes (CV) of Tris buffer (25 mM Tris pH 7.0, 150 mM NaCl, 5 mM EDTA, 2 mM sodium azide (NaN3)), 5 CVs of Triton X-114 buffer (25 mM Tris pH 7.0, 150 mM NaCl, 5 mM EDTA, 0.1% Triton X-114, 2 mM NaN3), 10 CVs of Tris buffer, 2 CVs of KP wash buffer (0.4 M potassium phosphate pH 7.0, 5 mM EDTA, 0.02% Polysorbate Tween 20), and finally 10 CVs of Tris buffer. Protein was eluted with 5 CVs of elution buffer (50 mM sodium citrate pH 3.0, 150 mM NaCl) and immediately neutralized with 1 M Tris pH 8.0 buffer. The neutralized elution was concentrated and purified over a HiLoad 16/600 Superdex 200 SEC column (GE Healthcare, #28–983-36) with arginine buffer (200 mM arginine, 137 mM succinic acid, 1 mM NaN3) as the mobile phase. Eluted fractions were collected in 1 mL increments and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The final eluant pool was concentrated and formulated into 20 mM histidine acetate (HisOAc) pH 5.5, 240 mM sucrose, 0.02% Polysorbate Tween 20.
Bivalent biepitopic antibodies
Bivalent biepitopic antibodies were produced using knob-into-hole CH3 variantsCitation36,Citation37 together with in vitro assembly of separately expressed half-antibodies as described previously.Citation38
r:Fab-IgG antibodies
r:Fab-IgG formatsCitation14 were constructed by genetically linking an additional heavy chain Fab region (VH-CH1) N-terminal to the native heavy chain using a 7 residue Gly-Ser linker (GGGGSGG). r:Fab-IgGs were produced in one of three formats. Monoepitopic versions are a two DNA system because the same light chain pairs with all four Fab region heavy chains. Thus, no additional engineering was required to control chain pairing, and accordingly monoepitopic r:Fab-IgGs were expressed and purified as described above for native IgGs.
Biepitopic versions of r:Fab-IgG are a three DNA system, and proper chain pairing was accomplished using distinct approaches for anti-OX40 and anti-DR5 formats. Anti-OX40 r:Fab-IgGs used a homodimeric heavy chain format (Ab1-Ab2 and Ab2-Ab1 in ) where chain pairing was controlled by variants in the Fab region as described previously.Citation39 VH/VL regions used orthogonal variant pairs VH-Q39K/VL-Q38E or VH Q39E/VL Q38K (Kabat numbering), and CH1/CL regions used orthogonal variant pairs CH1-S183E/CL-V133K or CH1-S183K/CL-V133E (EU numbering). Single heavy chain DNA and the two cognate light chain DNAs were co-transfected into CHO cells and r:Fab-IgGs were purified as described above for native IgGs.
Anti-DR5 r:Fab-IgGs used a heterodimeric heavy chain format (Ab1/Ab2 in ) that leveraged knob-into-hole CH3 variants together with in vitro assembly of separately expressed half-antibodies. Half-antibodies were expressed, purified, and assembled as described above for bivalent biepitopic antibodies.
c:Fab-IgG antibodies
Coupled Fab-IgG (c:Fab-IgG) anti-OX40 antibodies were constructed by conjugating full-length human IgG1 containing a free cysteine engineered at Ckappa position 149 [ThioMabTM(K149C)]Citation40,Citation41 to a Fab engineered with a C-terminal free cysteine on the heavy chain. ThioMabTM(K149C) antibodies were recombinantly expressed in CHO cells and purified as described previously.Citation42 Fabs contained a C-terminally extended variant hinge sequence on the heavy chain (DKTHTSPPC) that provided the free C-terminal cysteine for conjugation (referred to as Fab-C). Fab-C were expressed in E. coli and purified as previously described.Citation43 The chemical linker for all coupled formats used in this study was a bis-maleimido polyethylene glycol (BMPEG) linker containing 3 PEG units.
Fab-C were first functionalized with bis-maleimide crosslinker. Fresh bis-maleimide crosslinker (BMPEG) (ThermoFisher Scientific, 22337) was reconstituted to final concentration of 50 mM in N,N-dimethylacetamide (Sigma Aldrich, D5511). Purified Fab-C was then reacted with 10 mole equivalents of BMPEG at pH 5.0 for 2 h at room temperature, and reaction progress was monitored by mass spectrometry. After incubation for 2 h, the reaction was complete and excess crosslinker was removed by simple buffer exchange with 10,000 molecular weight cut-off (MWCO) Amicon Ultra-15 centrifugal filter units (UFC901096, Millipore) into conjugation buffer (25 mM sodium acetate (NaOAc) pH 5.0, 2 mM NaN3). Post buffer exchange, the final functionalized Fab (Fab-BMPEG) was analyzed with mass spectrometry.
c:Fab-IgG was then conjugated and purified. Purified ThioMabTM(K149C) antibody (5 mg/mL) was reacted with five molar excess of Fab-BMPEG. The reaction mixture was conditioned with conjugation buffer to a final pH of 5.0 and incubated at room temperature for 21 h. Reaction progress was monitored with mass spectrometry. After incubation for 21 h, the reaction was complete and the reaction mixture was purified with hydrophobic interaction chromatography (HIC). Briefly, the sample was conditioned with 1.5 M ammonium sulfate and loaded onto a ProPacTM HIC-10 column (5 μm, 7.8 mm x 75 mm) (Thermo Scientific, 063665). After sample load, the column was washed with 5 CVs of HIC-Buffer A (50 mM potassium phosphate pH 7.0, 1 M ammonium sulfate) and eluted with a linear gradient from 0% to 80% HIC-Buffer B (50 mM potassium phosphate pH 7.0, 20% isopropanol) over 60 CVs. Eluted fractions were collected and analyzed by liquid chromatography mass spectrometry (LC/MS) prior to formulation. The final conjugate was formulated into 20 mM HisOAc pH 5.5, 150 mM NaCl buffer and quality of final conjugate was assessed with LC/MS, SDS-PAGE, and SEC.
c:IgG-IgG antibodies
Coupled IgG-IgG (c:IgG-IgG) anti-OX40 antibodies were constructed by engineering an asymmetric ThiomAbTM(K149C) antibody in which only one of the half-antibodies has a free cysteine at Ckappa position 149. Half-antibodies were expressed, purified, and assembled as described above to form the knob-into-hole ThioMabTM(KiH, K149C). Due to the annealing process, the engineered cysteine on the ThioMabTM(KiH, K149C) antibody was blocked, and thus unreactive towards maleimide crosslinker. To remove the adduct, the antibody was reduced and reoxidized as previously described.Citation42 The final deprotected ThioMabTM(KiH, K149C) antibody was formulated into conjugation buffer and stored at 4°C.
Deprotected ThioMabTM(KiH, K149C) antibody was reacted with 10 mole equivalents of freshly prepared BMPEG (see above) at pH 5.0 for 2 h and reaction progress was monitored by mass spectrometry. After the reaction was completed, excess crosslinker was removed by ion exchange chromatography (IEX). Briefly, the reaction mixture was diluted 10-fold with IEX-Buffer A (25 mM NaOAc pH 5.0, 2 mM NaN3) and loaded onto a pre-equilibrated Hi-Trap SPHP (GE Healthcare, 17115201) column. After sample load, the column was washed with 5 CVs of IEX-Buffer A and eluted with a step gradient from 0% −100% over 1 CV with IEX-Buffer B (25 mM NaOAc pH 5.0, 1000 mM NaCl, 2 mM NaN3). Fractions were collected and analyzed by mass spectrometry. The final ThioMabTM(KiH, BMPEG) sample was concentrated to 5 mg/mL and formulated into conjugation buffer. The purified ThioMabTM(KiH, BMPEG) was then added to 1.5 molar excess of ThioMabTM(KiH, K149C) in the same buffer to form the coupled IgG. The reaction mixture was incubated at room temperature for 48 h and reaction progress was monitored with LC/MS analysis. After 48 h, the reaction was complete and the mixture was purified with HIC as described earlier. The final purified coupled IgG was formulated into 20 mM HisOAc pH 5.5, 150 mM NaCl.
Fab antibodies
Fab proteins for crystallographic experiments were expressed in E. coli and purified in two steps. First, cell lysate supernatant was loaded onto a G sepharose column and eluted with 0.6% acetic acid. Second, Fab-containing fractions were pooled and purified using a SP sepharose column (GE Healthcare). Loading buffer contained 2-(N-morpholino)ethanesulfonic acid (MES) at pH 5.5 and the protein was eluted on a NaCl gradient.
Production yield and purity
Production yields of recombinantly expressed antibodies and antibody formats, calculated as the final amount of purified protein (mg) divided by the number of liters (L) of transiently expressed CHO cells, were as follows: IgGs and ThioMabTM(K149C) IgGs (73–134 mg/L); knob half-antibody (63–116 mg/L); hole half-antibody (65–88 mg/L); RGY IgGs (30–65 mg/L); r:Fv-IgGs (37–88 mg/L); r:Fab-IgGs (47–86 mg/L). Production yield of Fab-C was 42–185 mg/L of E. coli culture. For formats that were in vitro assembled or conjugated, yield was calculated as the final amount of purified protein (mg) divided by the starting amount (mg) of total precursor IgG (Fab-C was non-limiting and used in excess). Yields were as follows: IgG Ab1/2 (40% from precursor knob + hole half-antibodies); c:Fab-IgG (26% from precursor ThioMabTM(K149C) IgG); c:IgG-IgG (22% from precursor bispecific ThioMabTM(K149C)).
Protein quality was assessed by SEC using a Waters xBridge BEH200A SEC 3.5um (7.8 x 300 mm) column (Waters,176003596). Molecular weight of all antibodies and proteins was confirmed by LC/MS. During purification, no major differences in the aggregation profiles were observed compared to native parental IgGs. All antibodies and antibody formats tested were purified to greater than 99% monomeric species, and remained monodisperse in solution after purification. No additional glycosylation or other post-translational modifications were observed in any of the engineered formats, including in the linkers, as determined by LC/MS.
Protein reagents
Human OX40 receptor extracellular domain (ECD, residues L29-D170 with an N-terminal His-tag) for crystallography was co-expressed in Sf9 insect cells with endoglycosidase H. OX40-containing medium was separated from cell debris by centrifugation. Protein was purified by first using a 10 mL Ni-NTA Superflow column (Qiagen). OX40 was eluted with buffer containing 20 mM tris(hydroxymethyl)aminomethane (Tris) at pH 8.0, 300 mM NaCl and 300 mM imidazole. Fractions containing OX40 were pooled and purified on an S-75 SEC column (Pharmacia) equilibrated in 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (Hepes) pH 7.2 and 200 mM NaCl. The His-tag was cleaved by overnight incubation with thrombin (Sigma) at 4°C. Cleaved OX40 protein was collected in the flow-through fraction of a 1.0 mL Ni-NTA Sepharose (Qiagen) column and was further purified by SEC on a S-75 column (Pharmacia) equilibrated in the same S-75 buffer mentioned above.
Human Apo2L ECD with an N-terminal Flag tag was expressed in E. coli. Cell paste was resuspended in cell lysis buffer and homogenized with a microfluidizer. Homogenized cell lysate was then loaded onto a 30 mL HisTrap column (GE Healthcare). The protein was then eluted from the column with buffer containing high concentration of imidazole. The eluate was directly loaded onto a 5 mL HiTrap SP HP column (GE Healthcare) in flowthrough mode to remove high molecular weight (HMW) contaminants. To enrich for trimeric FLAG-Apo2L, the flowthrough from the SP HP column was loaded onto a 50 mL ceramic hydroxyapatite (CHT) column (BioRad) followed by 12 mL POROS XS (Thermo Fisher). The purified protein was concentrated and formulated into 500 mM arginine succinate pH 7.2, 20 mM TrisCl, 1 mM TCEP buffer.
Affinity measurements
Solution affinity constants for binding to OX40 and DR5 were determined by surface plasmon resonance on a Biacore T200. Anti-OX40 antibody molecules, anti-DR5 antibody molecules, or Fc-OX40L (Acro Biosystem, OXL-H526x) were captured by Protein A chip. Series diluted OX40 (G&P Bioscience, FCL-2479) or FLAG-Apo2L in Biacore HBSP buffer was injected for 3 min, followed by 5 min dissociation. Affinity constants were obtained by kinetic fitting.
For cell binding assays, anti-OX40 antibody molecules and Fc-OX40L (Sino Biological, cat # 13127-H01H) were labeled with fluorescein using the Lightning-Link Antibody Labeling Kit (Novus Biologicals, Cat No. 707–0010). OX40+ Jurkat cells were washed twice with fluorescence-activated cell sorting (FACS) staining buffer (phosphate-buffered saline (PBS) containing 1% bovine serum albumin). 1 × 106 cells were then incubated with the indicated concentration of fluorescein-labeled antibody or ligand in 100 μL FACS staining buffer for 1 h at 4°C. The cells were then washed twice with 200 μL FACS staining buffer. Flow cytometry was performed on a BD FACS Celesta and data was analyzed using FlowJo. EC50 values were calculated based on fluorescein mean fluorescent intensity of gated live cells over a series of labeled protein concentrations.
Analytical SEC and SEC-MALS
SEC profiles of OX40 antibody and ligand complexes ()) were determined by SEC. Briefly, purified antibodies and human OX40 (G&P Biosciences FCL2479) were mixed in varying molar ratios in PBS. The data in the figure were from 1:1 ratio. 20 µl mixtures were injected and run 15 min at 0.3 ml/min using isocratic gradient over a TSKgel SuperSW3000 4 µm (4.6 mm ID x 30 cm) column analyzed by Thermo Scientific DIONEX UltiMate 3000 in 200 mM Potassium Phosphate/250 mM Potassium Chloride, pH 7.0 running buffer.
Molecular weights of OX40 antibody and ligand complexes ()) were determined by SEC linked in-line with Multiple Angle Light Scattering (SEC-MALS) as previously described.Citation44 Briefly, purified antibodies and human OX40 (G&P Biosciences FCL2479) were mixed in varying molar ratios in PBS. The mixtures were run over a Waters xBridge BEH200A SEC 3.5um (7.8 x 300 mm) column and analyzed by an Agilent 1200 HPLC connected to Wyatt Technology detectors DAWN HELEOS-II multiangle laser light scattering photometer and Optilab T-rEX differential refractive index detector.
Cell-based assays
For the OX40+ Jurkat reporter assay, cells were seeded at 80,000 cells/well in 20 µl AIM-V media (Thermo Fischer Scientific, Cat. # 12055–091) in 384-well tissue culture plate (Corning Inc., cat# 3985BC). OX40L ECD fused to the Fc region of human IgG1 (OX40L-Fc) was obtained from Sino Biological (cat # 13127-H01H). Extrinsic crosslinking of human IgG1 antibodies and OX40L-Fc (+XL in )) utilized goat anti-human IgG Fcγ fragment specific antibody (Jackson ImmunoResearch Laboratories Inc, cat# 109–005-098) at a final concentration of 10 µg/ml. Anti-OX40 antibodies or OX40L-Fc were serially diluted in AIM-V media at 2X concentration and 20 µl of the diluted antibodies were added to each well and incubated for 16–18 h in CO2 incubator with 37°C, 5% CO2., and 95% relative humidity. 40 µl Bright Glo (Promega cat# E2610) was added and mixed at room temperature for 10 min. Luminescence was detected on an Infinite M1000 Pro plate reader (Tecan).
For primary T cell assays, human CD4+ CD45RO+ memory T cells were isolated from buffy coat. L cells expressing CD80 (B7-1) and CD32a (FcγRIIa), or CD32a only were used as surrogate APCs. L cells (1 × 106 per mL) were irradiated at 5 × 103 Rad then seeded in Corning 96 well plate (Cat. # 3603) at 5 × 103 cells/50 μL in RPMI Growth Media (RPMI 1640, 10% heat inactivated fetal bovine serum (FBS), 100U penicillin/mL, 100 ug streptomycin/mL, 2 mM L-Glutamine). Soluble anti-CD3 antibody (mouse anti-human CD3 clone SP34 (BD Biosciences Cat# 557052) was prepared at 160 ng/mL in RPMI Growth Media, then 50 μL was added per well. Anti-OX40 antibodies were prepared at 1280 ng/mL followed by serial 4-fold dilutions, 5 steps for 6-points curve in RPMI Growth Media, then 50 μL was added per well. CD4+ memory T cells (50 μL at 50 × 103 cells/50 μL) were added to each well, and the plate with total volume of 200 μL per well was incubated for 7 days hours in CO2 incubator with 37°C, 5% CO2, and 95% relative humidity. 100 μL cell supernatant was removed per well, followed by addition of 100 μL CellTiter-Glo® (Promega, cat#G7573) per well. The plate was mixed at room temperature for 10 min. Luminescence was detected on an Infinite M1000 Pro plate reader (Tecan).
For anti-DR5 assays, human COLO 205 cells were maintained in RPMI medium supplemented with 1% L-glutamine and 10% heat-inactivated FBS (Rockland Immunochemicals Inc., Limerick, PA, USA) under 5% CO2 at 37°C. COLO 205 cells were plated at 100,000 cells/well of a 96-well white-walled plate (Corning) in 50 µl of growth media and allowed to adhere overnight. Dilutions of antibodies or FLAG-Apo2L protein were prepared in growth media, and 50 µl of each antibody sample was added to each well. For crosslinking of FLAG-Apo2L, anti-FLAG antibody (mouse anti-FLAG M2 antibody, Sigma) was used at 1:1 ratio at each concentration. For caspase-8 activity, cells were incubated with the antibody for 4 h prior to the addition of Caspase-Glo 8 (Promega). For cell viability, cells were incubated with the antibody for 24 h prior to the addition of CellTiter-Glo (Promega). Experiments were done in triplicate and luminescence was read using Envision (PerkinElmer).
Flow cytometry
For FACS experiments, primary human CD4+ memory T cells were co-cultured with B7-1− or B7-1+ L cells. At each time point, cells were stained with fluorochrome-conjugated anti-human OX40 (clone ACT35; PE-CF594) and anti-human CD4 (clone SK3; BV650). The percentage of CD4+ T cells expressing OX40 was analyzed by multicolor-flow cytometry (triplicate of two healthy donors).
Microscopy
Primary T cells were allowed to settle on polylysine-coated (Sigma P4704) wall-less 96-well plates (Curiox 96-CC-TC-05) for 1 h on ice in 10 µl carbonate-free medium (Gibco 18045–088, containing 10% FBS, 1% L-glutamine, 10 mM Hepes and 1% pen/strep) at 15,000/well for 3 day-stimulated (on anti-CD3-coated dishes with soluble anti-CD28) and 30,000/well for unstimulated. Then, 10 µl of 2x anti-OX40 antibodies or anti-gD isotype control were added to a final concentration of 6.7 nM and incubated on ice (to prevent internalization and trafficking) for 1 h. Plates were then washed, cells fixed with 3% paraformaldehyde, quenched with 50 mM NH4Cl and permeabilized with saponin buffer as previously described.Citation45 Bound antibodies were detected with Cy3-conjugated donkey F(ab’)2 anti-human H&L (Jackson Immunoresearch 709–166-149, 1:800). Coverslips were mounted in Vectashield with DAPI (Vector Labs H-1200, 6.6 µl per well) and plates imaged by immunofluorescence microscopy on a Zeiss AxioM2 microscope at 63x, as published.Citation45
Crystallographic structure solution
For the generation of the binary OX40:Fab1 and OX40:Fab2 complexes, 2 to 1 molar ratio amounts of OX40 ECD and Fab1 or Fab2 were mixed together and incubated overnight at 4°C. For ternary OX40:Fab1:Fab2 complex, equimolar amounts of OX40 ECD, Fab1, and Fab2 were mixed together and incubated overnight at 4°C. Each of the binary and ternary complexes were then purified by SEC on a S200 16/60 column (GE Healthcare) equilibrated in 25 mM Tris pH 7.5 and 0.15 M NaCl. Samples were analyzed by SDS-PAGE and LC/MS, which revealed the required components in the HMW peak fractions of the final SEC step. The HMW peak fractions were concentrated to 10 mg/mL using a Vivaspin concentrator with 10,000 MWCO.
Crystallization trials were performed using sitting-drop vapor diffusion method with commercially available sparse-matrix screens in 96-well format. The final crystallization condition for the binary OX4:Fab1 complex contained 73% 2-methyl-2,4-pentanediol and 100 mM HEPES pH 7.5. The binary OX40:Fab2 complex crystallization condition contained 45% PEG 300, 100 mM phosphate citrate pH 4.2. The ternary OX40:Fab1:Fab2 complex final crystallization condition contained 0.1 M Tris pH 8.5, 1.5 M D-L malic acid pH 7.0 and 0.15 mM dimethylethylammonium propane sulfonate (also known as NDSB-195). Crystals of the binary complexes were immersed into liquid nitrogen directly from the drops, while the ternary complex was preserved for data collection by brief soaking in a cryo-protectant buffer (25% glycerol added to the reservoir solution) followed by rapid immersion into liquid nitrogen.
Diffraction data were collected at the Advanced Light Source beamline 5.0.2 for binary OX40:Fab1 complex and ternary OX40:Fab1:Fab2 complex, while data for OX40:Fab2 were collected at the Stanford Synchrotron Radiation Lightsource beam line 12–2. Data reduction was performed using XDS.Citation46 The structure was solved by molecular replacement (MR) using PhaserCitation47 with an OX40 model from PDB 2HEVCitation23 and a prior OX40:Fab2 binary complex structure we had determined. As the Fab components were not forthcoming in the initial solutions, the elbow angles for the two Fab molecules were explored using rotation searches against a series of altered elbow-angle Fab structure models generated based on PDB 1FVD. Models of elbow angles at +70° and 0° gave the highest signal-to-noise ratio (3x). A full MR search was then performed using these two best elbow angle models (+70°, 0° elbow angles) with the previously determined OX40:Fab2 complex structure that had the constant domain (CH1 and CL) of the Fab removed. Inspection of the electron density suggested the Fab model with 0° elbow angle gave the best solution and corresponded to the Fab of Ab1. The ternary complex was iteratively built with COOTCitation48 and refined using PhenixCitation49 and BUSTERCitation50 (BUSTER version 2.11.5. Cambridge, United Kingdom: Global Phasing Ltd). Final refinement statistics are shown in . All structures have been deposited in the Protein Data Bank with accession codes as follows: ternary complex OX40:Fab1:Fab2 (6OGX), binary complex OX40:Fab1 (6OKN), and binary complex OX40:Fab2 (6OKM). Structural figures were prepared with PyMOL (Schrodinger LLC).
Animal study oversight
All animal studies were reviewed and approved by Genentech’s Institutional Animal Care and Use Committee.
In Vivo pharmacokinetic studies
PK of tetravalent antibody formats and relevant controls was tested in C57BL-6 or C.B-17 SCID mice. PK studies were performed with single intravenous (i.v) dose of 10 mg/kg of antibodies, and serum samples (n = 3/time point/group) were collected at various time points after dosing for PK analysis out to Day 21 post-dose.
A sandwich ELISA with a colorimetric detection system was used to quantitate antibody serum levels. Microtiter plates were coated with sheep anti-human IgG to capture antibodies. Diluted serum samples, standards, and controls were added to the plate and incubated. Subsequently, goat anti-human IgG-horseradish peroxidase (Jackson ImmunoResearch; Cat. No. 109–035-088) was added for detection and incubated. Peroxidase substrate (tetramethyl benzidine) was added to develop color, and the reaction was stopped by adding 1 M phosphoric acid. The plates were read at 450 nm for detection absorbance and 620 nm for reference absorbance. Sample concentration was determined by entering data into a four-parameter logistic curve-fitting program. The reporting range for the assay was 0.156–20 ng/mL anti-OX40 antibody. The minimum sample dilution was 1/100, resulting in a minimum quantifiable concentration of 15.6 ng/mL for mouse serum.
In Vivo pharmacodynamic studies
Human OX40 knockin (hOX40ki) C57BL/6N mice were generated at Genentech. In contrast to WT mice that express mouse OX40 on activated CD4+ and CD8+ T cells, human OX40 was only expressed on CD4+, but not on CD8+ T cells in hOX40ki mice (Supplementary Figure 6). PD studies measured T cell activation in response to immunization with KLH antigen. hOX40ki mice were subcutaneously (tail base) injected with PBS or 50 μg Imject mcKLH Subunits (ThermoFisher Scientific; Cat. No. 77649) emulsified in complete Freund’s adjuvant (CFA) H37 Ra (BD Difco; Cat. No. 231131) at 1:1 ratio for a total volume of 100 μL per mouse. On Day 1, animals were grouped and intravenously administered 10 mg/kg of anti-human OX40 human IgG1 antibodies or anti-gD isotype control.
To quantify the anti-KLH IgG antibody responses, mice from different groups were bled on Day 14 and serum anti-KLH IgG levels were measured by ELISA kit according to the manufacturer’s instruction (Life Diagnostics, Inc; Cat. No. KLHG-1). To access memory responses, mice were intraperitoneally rechallenged on Day 16 with 50 μg soluble KLH and draining lymph nodes were profiled 4 days post-rechallenge (Day 20). To assess the activation profile of T cells, draining LN cells were stained with fluorochrome conjugated anti-CD4, CD8, CD44, and CD62L antibodies and analyzed by multicolor-flow cytometry. CD44 and CD62L were used to determine the central memory (CM; CD44+CD62L+) and effector memory (EM; CD44+CD62L−) phenotype of CD4+ and CD8+ T cells. Draining LN cells were also stimulated in vitro with 10 μg/mL soluble KLH. For the results described in –i) the experiment used 105 cells/well, whereas for the results described in –l) the experiment used 106 cells/well. After 5 days, supernatants were collected and IFN-γ production was assayed using Luminex.
Abbreviations
Author contributions
Y.Y., W.L.M., E.C., J.Q.H., and G.A.L. performed antibody design. Y.Y. and W.L.M. performed molecular biology. S.H.Y., W.L.M., B.L., J.Z., and H.C. designed and performed in vitro assays. S.M., P.P.S., and R.C. designed and performed in vivo mouse experiments. C.G. and E.C. conjugated, assembled, and purified proteins. M.U. and G.d.L.B. crystallized the Fab/Fab/OX40 complex. M.U. and S.F.H. determined and refined the crystallographic structure. S.J.S. performed microscopy. S.M., A.A., and R.J.B. contributed ideas and guided experiments. S.S. designed and performed PK experiments. J.M.K. and G.A.L. developed the concept and led the research. G.A.L. wrote the manuscript. All authors analyzed data, discussed results, and contributed to or commented on the writing of the manuscript.
Disclosure of Potential Conflicts of Interest
All authors are current or former employees of Genentech, a member of the Roche Group, and are shareholders in Roche. This study was supported by internal Genentech funds, and the funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplemental Material
Download Zip (27.6 MB)Acknowledgments
We thank Peter Luan, Ingrid Kim, Sharon Viajar, May Lin, Chris Cox, and John Tran for technical contributions. Portions of this research were carried out at the Advanced Light Source (ALS) and the Stanford Synchrotron Radiation Lightsource (SSRL), which are supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contracts DE-AC02-05CH11231 (ALS) and DE-AC02-76SF00515 (SSRL). The SSRL Structural Molecular Biology Program is further supported by the DOE Office of Biological and Environmental Research, and by the NIH, National Institute of General Medical Sciences (including P41GM103393).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
Related Research Data
References
- Croft M, Benedict CA, Ware CF. Clinical targeting of the TNF and TNFR superfamilies. Nat Rev Drug Discov. 2013;12:147–68. doi:10.1038/nrd3930.
- Faustman DL, Davis M. TNF receptor 2 and disease: autoimmunity and regenerative medicine. Front Immunol. 2013;4:478. doi:10.3389/fimmu.2013.00478.
- Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discov. 2018;17:509–27. doi:10.1038/nrd.2018.75.
- Sturgill ER, Redmond WL. TNFR agonists: a review of current biologics targeting OX40, 4-1BB, CD27, and GITR. Am J Hematol Oncol. 2017;13:4–15.
- Vanamee ES, Faustman DL. Structural principles of tumor necrosis factor superfamily signaling. Sci Signal. 2018;11:eaao4910. doi:10.1126/scisignal.aao4910.
- Wajant H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 2015;22:1727–41. doi:10.1038/cdd.2015.109.
- Furness AJ, Vargas FA, Peggs KS, Quezada SA. Impact of tumour microenvironment and Fc receptors on the activity of immunomodulatory antibodies. Trends Immunol. 2014;35:290–98. doi:10.1016/j.it.2014.05.002.
- Kim JM, Ashkenazi A. Fcgamma receptors enable anticancer action of proapoptotic and immune-modulatory antibodies. J Exp Med. 2013;210:1647–51. doi:10.1084/jem.20130166.
- Wilson NS, Yang B, Yang A, Loeser S, Marsters S, Lawrence D, Li Y, Pitti R, Totpal K, Yee S, et al. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011;19:101–13. doi:10.1016/j.ccr.2010.11.012.
- Ashkenazi A. Targeting the extrinsic apoptotic pathway in cancer: lessons learned and future directions. J Clin Invest. 2015;125:487–89. doi:10.1172/JCI80323.
- Bremer E. Targeting of the tumor necrosis factor receptor superfamily for cancer immunotherapy. ISRN Oncol. 2013;2013:371854.
- Chester C, Ambulkar S, Kohrt HE. 4-1BB agonism: adding the accelerator to cancer immunotherapy. Cancer Immunol Immunother. 2016;65:1243–48. doi:10.1007/s00262-016-1829-2.
- Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS, Levy R. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med. 2018;10.
- Miller K, Meng G, Liu J, Hurst A, Hsei V, Wong WL, et al. Design, construction, and in vitro analyses of multivalent antibodies. J Immunol. 2003;170:4854–61. doi:10.4049/jimmunol.170.9.4854.
- Wu C, Ying H, Grinnell C, Bryant S, Miller R, Clabbers A, et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotechnol. 2007;25:1290–97. doi:10.1038/nbt1276.
- Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343:1260–63. doi:10.1126/science.1248943.
- Zhang D, Goldberg MV, Chiu ML. Fc engineering approaches to enhance the agonism and effector functions of an Anti-OX40 antibody. J Biol Chem. 2016;291:27134–46. doi:10.1074/jbc.M116.757773.
- Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29:457–66. doi:10.1093/protein/gzw039.
- Zhang D, Armstrong AA, Tam SH, McCarthy SG, Luo J, Gilliland GL, et al. Functional optimization of agonistic antibodies to OX40 receptor with novel Fc mutations to promote antibody multimerization. MAbs. 2017;9:1129–42. doi:10.1080/19420862.2017.1358838.
- Walker LS, Gulbranson-Judge A, Flynn S, Brocker T, Raykundalia C, Goodall M, et al. Compromised OX40 function in CD28-deficient mice is linked with failure to develop CXC chemokine receptor 5-positive CD4 cells and germinal centers. J Exp Med. 1999;190:1115–22. doi:10.1084/jem.190.8.1115.
- Hunig T. The storm has cleared: lessons from the CD28 superagonist TGN1412 trial. Nat Rev Immunol. 2012;12:317–18. doi:10.1038/nri3192.
- Tacke M, Hanke G, Hanke T, Hunig T. CD28-mediated induction of proliferation in resting T cells in vitro and in vivo without engagement of the T cell receptor: evidence for functionally distinct forms of CD28. Eur J Immunol. 1997;27:239–47. doi:10.1002/eji.1830270136.
- Compaan DM, Hymowitz SG. The crystal structure of the costimulatory OX40-OX40L complex. Structure. 2006;14:1321–30. doi:10.1016/j.str.2006.06.015.
- Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J. 2012;14:296–302. doi:10.1208/s12248-012-9340-y.
- Horenstein AL, Chillemi A, Quarona V, Zito A, Mariani V, Faini AC, et al. Antibody mimicry, receptors and clinical applications. Hum Antibodies. 2017;25:75–85. doi:10.3233/HAB-160305.
- Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov. 2018;17:197–223. doi:10.1038/nrd.2017.227.
- Dempke WCM, Fenchel K, Uciechowski P, Dale SP. Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur J Cancer. 2017;74:55–72. doi:10.1016/j.ejca.2017.01.001.
- Li JY, Perry SR, Muniz-Medina V, Wang X, Wetzel LK, Rebelatto MC, et al. A biparatopic HER2-targeting antibody-drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2-targeted therapy. Cancer Cell. 2016;29:117–29. doi:10.1016/j.ccell.2015.12.008.
- Kundig TM, Shahinian A, Kawai K, Mittrucker HW, Sebzda E, Bachmann MF, et al. Duration of TCR stimulation determines costimulatory requirement of T cells. Immunity. 1996;5:41–52. doi:10.1016/S1074-7613(00)80308-8.
- Jiang J, Liu C, Liu M, Shen Y, Hu X, Wang Q, et al. OX40 signaling is involved in the autoactivation of CD4(+)CD28(-) T cells and contributes to the pathogenesis of autoimmune arthritis. Arthritis Res Ther. 2017;19:67. doi:10.1186/s13075-017-1261-9.
- Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–30. doi:10.1038/nature21349.
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–33. doi:10.1126/science.aaf1292.
- Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–27. doi:10.1126/science.aaf0683.
- Fecher LA, Agarwala SS, Hodi FS, Weber JS. Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist. 2013;18:733–43. doi:10.1634/theoncologist.2012-0483.
- Eaton DL, Wood WI, Eaton D, Hass PE, Hollingshead P, Wion K, et al. Construction and characterization of an active factor VIII variant lacking the central one-third of the molecule. Biochemistry. 1986;25:8343–47. doi:10.1021/bi00374a001.
- Atwell S, Ridgway JB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol. 1997;270:26–35. doi:10.1006/jmbi.1997.1116.
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9:617–21. doi:10.1093/protein/9.7.617.
- Spiess C, Merchant M, Huang A, Zheng Z, Yang NY, Peng J, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat Biotechnol. 2013;31:753–58. doi:10.1038/nbt.2595.
- Dillon M, Yin Y, Zhou J, McCarty L, Ellerman D, Slaga D, et al. Efficient production of bispecific IgG of different isotypes and species of origin in single mammalian cells. MAbs. 2017;9:213–30. doi:10.1080/19420862.2016.1267089.
- Pillow TH, Sadowsky JD, Zhang D, Yu SF, Del Rosario G, Xu K, et al. Decoupling stability and release in disulfide bonds with antibody-small molecule conjugates. Chem Sci. 2017;8:366–70. doi:10.1039/c7sc01787a.
- Vollmar BS, Wei B, Ohri R, Zhou J, He J, Yu SF, et al. Attachment site cysteine Thiol pKa is a key driver for site-dependent stability of THIOMAB antibody-drug conjugates. Bioconjug Chem. 2017;28:2538–48. doi:10.1021/acs.bioconjchem.7b00365.
- Sadowsky JD, Pillow TH, Chen J, Fan F, He C, Wang Y, et al. Development of efficient chemistry to generate site-specific disulfide-linked protein- and peptide-payload conjugates: application to THIOMAB antibody-drug conjugates. Bioconjug Chem. 2017;28:2086–98. doi:10.1021/acs.bioconjchem.7b00258.
- Scheer JM, Sandoval W, Elliott JM, Shao L, Luis E, Lewin-Koh S-C, Schaefer G, Vandlen R, Verma C. Reorienting the Fab domains of trastuzumab results in potent HER2 activators. PLoS One. 2012;7:e51817. doi:10.1371/journal.pone.0051817.
- Shatz W, Hass PE, Mathieu M, Kim HS, Leach K, Zhou M, Crawford Y, Shen A, Wang K, Chang DP, et al. Contribution of antibody hydrodynamic size to vitreal clearance revealed through rabbit studies using a species-matched Fab. Mol Pharm. 2016;13:2996–3003. doi:10.1021/acs.molpharmaceut.6b00345.
- Ingle GS, Scales SJ. DropArray, a wall-less 96-well plate for uptake and immunofluorescence microscopy, confirms CD22 recycles. Traffic. 2014;15:255–72. doi:10.1111/tra.12162.
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–32. doi:10.1107/S0907444909047337.
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–74. doi:10.1107/S0021889807021206.
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi:10.1107/S0907444910007493.
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi:10.1107/S0907444909052925.
- Dsafa Bricogne G, Blanc E, Brandl M, Flensburg C, Keller PWP, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, et al. BUSTER Version 2.11.5. Cambridge (UK): Global Phasing Ltd; 2016