
ABSTRACT
Phagocytosis plays important roles both in homeostasis and under pathological conditions. Fcγ receptor-mediated phagocytosis has been exploited as an integral mechanism for antibody-based therapies. Unlike Fcγ receptor-mediated phagocytosis, MerTK-mediated phagocytic clearance is immunologically silent. Here, we describe a bispecific antibody approach to harness MerTK for targeted clearance without inducing proinflammatory cytokine release associated with Fcγ receptor engagement. We generated bispecific antibodies targeting live B cells or amyloid beta aggregates to demonstrate the feasibility and versatility of this new approach.
Macrophages are professional phagocytes with a versatile ability to dispose various targets, such as apoptotic cells, pathogens and cancer cells. They express three different activating Fcγ receptors (FcγR), FcγRI, FcγRIIA and FcγRIIIA, that contain immunoreceptor tyrosine-based activation motifs (ITAM), and one inhibitory receptor, FcγRIIB, that has an immunoreceptor tyrosine-based inhibitory motif (ITIM). IgG-opsonized targets cross-link the activating FcγR in macrophages, which results in phosphorylation of tyrosine residues in ITAMs and recruitment of spleen tyrosine kinase (SYK) to activate downstream signaling events. Besides phagocytosis, engagement of activating FcγR can lead to the production of pro-inflammatory cytokines.Citation1 The release of pro-inflammatory cytokines triggered by therapeutic monoclonal antibodies can be a desired outcome for treating cancers. For other disease indications, however, such an effect may limit the therapeutic benefit due to the detrimental side-effects, such as neurotoxicity, vasogenic edema and cytokine release syndrome.Citation2–Citation5
The Tyro3-Axl-MerTK (TAM) family of receptor tyrosine kinases are important for maintaining tissue homeostasis. Aided by bridging molecules such as Gas6 and Protein S, MerTK-expressing macrophages recognize phosphatidylserine (PtdSer) expressed by apoptotic cells and remove them via a process termed efferocytosis.Citation6 Timely removal of apoptotic cells is critical for preventing auto-immunity and inflammation. Genetic ablation of MerTK causes lupus-like disease in aged mice.Citation7 Furthermore, MerTK has been proposed to promote anti-inflammatory functions through various mechanisms, including suppression of TLR mediated cytokine production,Citation8 inhibition of NF-κB signaling,Citation9 and synthesis of inflammation resolution mediators.Citation10 Thus, the anti-inflammatory feature of MerTK-mediated phagocytosis offers an opportunity for developing new antibody therapeutics that may avoid the pro-inflammatory effect associated with engaging activatory Fcγ receptors. For instance, IgG1 antibodies targeting amyloid beta (Aβ) in patients with Alzheimer’s disease have been investigated clinically. These antibodies are designed to promote the clearance of amyloid plaques by FcγR-expressing microglial cells.Citation11 However, in clinic trials, amyloid-related imaging abnormalities (ARIA) are dose-limiting neuroimaging findings associated with such antibodies. Thus, harnessing MerTK agonism may overcome such hurdles in clinical development. Similar benefits may also be highly desirable when developing antibody drugs for treating other neurodegenerative diseases associated with protein aggregates.
We envisioned that a MerTK antibody with agonistic potential could be used for further antibody engineering to trigger target-dependent activation of MerTK. We screened a panel of rabbit-derived MerTK monoclonal antibodies that were either produced as rabbit IgGs or reformatted into murine IgG2a LALAPG, which carries mutations (L234A, L235A and P329G) in the Fc region to disrupt binding to Fcγ receptors. Citation12,Citation13 In a cell-based assay, MerTK activation was measured by the increased phospho-AKT (Ser473) in human primary macrophage cells (). In addition, we used a MerTK small molecule inhibitor (SMI) to verify the MerTK-dependency and rule out possible non-target effect. By this assay, we identified a few positive MerTK antibodies that exhibited MerTK-dependent agonistic activity.
Figure 1. Identifying and characterizing MerTK agonist antibodies for engineering anti-CD20/MerTK bispecific antibodies capable of inducing phagocytosis of B cells by macrophages (a) Characterization of lead MerTK agonist antibodies measured by their ability to induce pAKT (Ser-473) in human MerTK+ macrophages using HTRF assay (Mean ± S.D of 2 technical replicates). Gas6-Fc is a positive control for MerTK activation. (b) Schematic depiction of B cell-dependent engagement of MerTK via anti-CD20/MerTK bispecific antibody. (c) Imagestream analysis of phagocytosis of live Raji cells by human primary macrophages in the presence of indicated bispecific antibody. A representation of Imagestream analysis is shown. Quantification is also presented (Mean ± S.D of one replicate each from 3 donors). (d-f) Flow cytometric analysis of phagocytosis of live Raji cells by human primary macrophages in the presence of indicated bispecific antibody, inhibitors and blocking agents (Mean ± S.D of one replicate each from 3 donors) (g) Activation of MerTK (phosphotyrosine) by the bispecific antibody in the presence of target Raji cells. Results are from using macrophages derived from two different human donors. See Supplementary Figure 5 for extended western blot. (h-i) TNF and IL-6 production by human primary macrophages stimulated with indicated plate coated antibodies. (Mean ± S.D of one replicate each from three different donors)
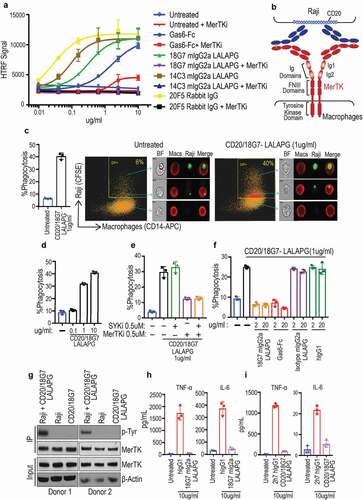
As a proof-of-principle of exploring MerTK agonism for targeted phagocytosis, we started with two MerTK antibodies (18G7 and 14C3) to generate CD20-targeting bispecific antibodies using the knob-hole technology.Citation14,Citation15 While the CD20/MerTK bispecific antibodies have a configuration of monovalent binding to CD20 and MerTK (), we anticipated that the clustering of the bispecific antibodies by CD20 expressing cells could trigger the activation of MerTK expressed by the phagocytes. To reduce binding to FcγRs, the bispecific antibodies were produced as a human IgG1 with N297G mutation (CD20/MerTK-N297G).Citation13,Citation16 We confirmed the binding of CD20/MerTK bispecific antibodies to MerTK-expressing human monocyte-derived macrophages and CD20+ Raji cells (Figure S1A). We then tested their activities in a phagocytosis assay using human macrophages as phagocytes and Raji cells as target cells. We found that the CD20/MerTK-N297G antibodies were able to induce macrophage phagocytosis of Raji cells (Figure S1B and 1C). The antibody-mediated phagocytosis is target specific because the MerTK bispecific antibody, CD20/18G7-N297G, failed to promote the uptake of CD20-negative Jurkat cells by macrophages (Figure S1D).
The observed phagocytosis in the presence of CD20/MerTK bispecific antibodies could be due to antibody-mediated “tethering” of target cells to macrophages.Citation17 In order to investigate the importance of kinase-dependent MerTK signaling, we performed a phagocytosis assay in the presence of a MerTK SMI (Figure S1B and C). As a control, we used a SMI to inhibit SYK, which is involved in activating FcγR downstream signaling. The specificity and potency of the inhibitors were verified in a MerTK-dependent efferocytosis assay using apoptotic Jurkat cells as the target cells (Figure S1E). In the phagocytosis assay using Raji cells and human macrophages, we found that the MerTK SMI partially reduced the phagocytosis mediated by two independent CD20/MerTK bispecific antibodies, CD20/18G7-N297G and CD20/14C3-N297G. Interestingly, phagocytosis was also decreased by the SYK inhibitor (Figure S1B and C), suggesting that the N297G mutation does not completely abolish the engagement of FcγR.13 In comparison, the combination of MerTK and SYK inhibitors completely blocked the phagocytosis, demonstrating that both FcγR and MerTK contribute to the phagocytosis triggered by the CD20/MerTK-N297G bispecific antibodies (Figure S1B and C).
To separate the effect of MerTK from FcγR, we produced a new CD20/MerTK bispecific hIgG1 carrying the LALAPG mutation, CD20/18G7-LALAPG.Citation12,Citation13,Citation18 The LALAPG and N297G bispecific antibodies exhibited similar binding to human primary macrophages and Raji cells (Figure S1F). CD20/18G7-LALAPG induced phagocytosis of live Raji cells by macrophages as assessed through Imagestream analysis (). Further, we found that phagocytosis of Raji cells by macrophages was dose dependent (). Unlike CD20/18G7-N297G, the phagocytic activity of CD20/18G7-LALAPG was reduced by the MerTK inhibitor, but not SYK inhibitor (). Additionally, we used a monospecific MerTK antibody (18G7 mIgG2a-LALAPG), or Gas6-Fc, to compete against the bispecific antibody. Both agents abolished the activity of CD20/18G7-LALAPG, further demonstrating the dependence on MerTK signaling (). To further establish that MerTK is activated by CD20/18G7-LALAPG through target mediated clustering, we co-cultured Raji and human macrophages with CD20/18G7-LALAPG for 30 minutes, and then activation of MerTK was assessed by phosphorylation of tyrosine residues. We observed robust activation of MerTK in macrophages only in the presence of both Raji (target) and the CD20/18G7-LALAPG ().
To determine the effect of MerTK activation on the release of pro-inflammatory cytokines, we incubated human primary macrophages with plate-coated monospecific MerTK agonist 18G7 mIgG2a-LALAPG (Figure S2A), or FcγR agonist hIgG1 for 24 hours. We found that activation of FcγR, but not MerTK, induced a robust production of pro-inflammatory cytokines, including tumor necrosis factor (TNF), and interleukin (IL)-6 ( and Figure S2A). Additionally, only FcγR activation increased the production of IL-1α, IL-1β, IFN-γ, IL-8, IP-10, RANTES, MIP-1α, MIP-1β, and IL-10 (Figure S2B). Similar results were obtained when we compared the bispecific MerTK agonist CD20/18G7 hIgG1-LALAPG with the anti-CD20 2h7 hIgG1 (FcγR agonist) ( and Figure S2C). Together, these findings support the feasibility of targeted phagocytosis that exclusively relies on immunologically silent MerTK signaling.
Antibody therapy has also been used for clearance of protein aggregates such as amyloid plaques.Citation11 This prompted us to explore the possibility of using MerTK bispecific antibody for targeted removal of protein aggregates. We chose aggregated Aβ as a target for this study. Clone 20F5, another anti-MerTK antibody identified in the agonist screening, cross-reacted with both human and mouse MerTK (Figure S4A, B). In addition, as a rabbit IgG, it exhibited MerTK-dependent agonist activity in both human () and mouse macrophages (Figure S3A). Using the knob-hole technology, we generated bispecific antibodies by pairing anti-MerTK 20F5 and GP120 (control) with anti-Aβ (clone 3D6).Citation19 To increase the binding to Aβ, we chose a “2 + 1” configuration in which the anti-Aβ arm is a stacked Fab-IgG (Figure S3B). The resulting bispecific antibodies 3D6/20F5-LALAPG and 3D6/GP120-LALAPG exhibited similar binding to immobilized Aβ (1–42) compared to 3D6 mIgG2a (Figure S3C). We used mouse bone marrow-derived macrophages (BMDMs) as phagocytes and tested the ability of 3D6/20F5-LALAPG to induce uptake of aggregated Aβ. Indeed, 3D6/20F5-LALAPG was able to trigger the phagocytosis of aggregated Aβ by BMDMs in a dose- and time-dependent manner compared to control 3D6/GP120-LALAPG (). We found that the uptake of aggregated Aβ was greatly reduced in the presence of anti-MerTK 20F5 rabbit IgG or a small molecule MerTK inhibitor (). Furthermore, unlike WT BMDMs, MerTK −/-BMDMs were unable to uptake aggregated Aβ (, Figure S3D). Finally, FcγR-engaging antibody 3D6 mIgG2a, but not anti-Aβ bispecific antibody 3D6/20F5 mIgG2a-LALAPG, induced the production of pro-inflammatory cytokine TNF ().
Figure 2. Anti-Aβ/MerTK bispecific antibody induces uptake of aggregated Aβ by BMDMs or microglia (a) Antibody dose-dependent uptake of aggregated Aβ by mouse BMDMs (Mean ± S.D of 3 technical replicates). (b) Time course of aggregated Aβ uptake by mouse BMDMs (Mean ± S.D of 3 technical replicates). Aggregated Aβ at 1 μM with antibodies at 10 ug/ml concentration. (c-e) BMDM uptake of aggregated Aβ induced by anti-Aβ/MerTK bispecific antibody is MerTK-dependent, as demonstrated by competition with the parental anti-MerTK antibody (20F5 Rabbit IgG) (c), inhibition by a MerTK SMI (d), or loss of activity in MerTK −/- BMDMs (e) (Mean ± S.D of 3 technical replicates). Aggregated Aβ at 1 μM, and antibodies at 10 ug/ml concentration. (f) TNF production by BMDMs stimulated with 3D6 mIgG2a (FcγR agonist) or 3D6/20F5 mIgG2a-LALAPG (MerTK agonist) (Mean ± S.D of 3 technical replicates). (g-i) Microglial cell uptake of aggregated Aβ induced by anti-Aβ/MerTK bispecific antibody is MerTK-dependent, as demonstrated by competition with the parental anti-MerTK antibody (20F5 Rabbit IgG) (g), inhibition by a MerTK SMI (h), or loss of activity in MerTK −/- microglia (i) (Mean ± S.D of 3 technical replicates). (j) TNF production by microglia stimulated with 3D6 mIgG2a (FcγR agonist) and 3D6/20F5 mIgG2a-LALAPG (MerTK agonist) and 3D6/GP120 mIgG2a-LALAPG (control) (Mean ± S.D of 3 technical replicates)
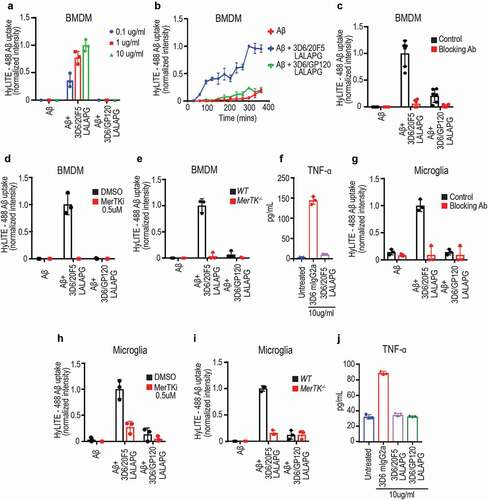
Microglial cells are specialized resident macrophages present in the central nervous system (CNS). They are the primary innate immune effector cells of the CNS, and are important for maintaining CNS homeostasis. Similar to the peripheral tissue resident macrophages, microglia also express MerTK.Citation20,Citation21 We found that 3D6/20F5-LALAPG induced Aβ aggregate uptake by primary mouse microglial cells. Similar to what were observed with BMDMs, the uptake of Aβ aggregate was MerTK dependent (–i). Additionally, we observed the production of pro-inflammatory cytokine TNF by microglia only in the presence of the FcγR-engaging 3D6 mIgG2a, but not with the MerTK-targeting bispecific antibody 3D6/20F5 mIgG2a-LALAPG (, and Figure S3E).
Here, we showed that anti-MerTK bispecific antibodies were able to mediate targeted phagocytosis of live cells or protein aggregates. Importantly, the MerTK-dependent signaling did not result in the release of pro-inflammatory cytokines. This is a major distinction from existing anti-Aβ antibodies, which rely on FcγR-mediated phagocytosis for efficacy. In in vitro studies, these antibodies induce the release of pro-inflammatory cytokines and activation of microglia upon cross-linking of FcγR,Citation2,Citation11,Citation19 which could contribute to the observed side-effects such as edema and micro-hemorrhage of the cerebral vasculature.Citation4,Citation5 In comparison, our approach of engaging MerTK signaling offers a therapeutic advantage by segregating phagocytosis from pro- inflammatory response. This is particularly important for treating diseases in which the pathological conditions could be worsened by inflammation. Given these promising results, future work will focus on optimizing the antibody format that would allow further investigation in disease-relevant models.
Materials and methods
Mertk and SYK inhibitors
The Axl/MerTK specific kinase inhibitor was described in patent WO 2015068767. SYK inhibitor, PRT062607 is obtained from selleckchem, Catalog S8032.
Mertk antibodies
Anti-MerTK antibodies were generated from New Zealand White rabbits immunized with recombinant murine (E23-S496) and human (R26-A499) MerTK proteins as described previously.Citation22 Briefly, B cell clones were selected based on binding to purified MerTK by enzyme-linked immunosorbent assay (ELISA) and MerTK-expressing cells by fluorescence-activated cell sorting (FACS). Variable regions of the light chain and heavy chain of positive clones were amplified by PCR and cloned into expression vectors. Recombinant antibodies were expressed in Expi293 cells, and purified with protein A. Antibody clones18G7, 14C3 and 20F5 were independent clones with distinct variable domain sequences.
Bispecific antibodies
Bispecific antibodies are produced in knobs-into-holes format as previously described.Citation14 Anti-CD20/MerTK are generated as full-length hIgG1 with either N297G or LALAPG mutations. Anti-Aβ/MerTK are full-length mIgG2a with LALAPG mutation. The CD20 arm is 2H7 Citation15 whereas the Aβ arm is 3D6.Citation19
Affinity determination
To measure the binding affinity of MerTK agonistic antibody, a surface plasmon resonance BIAcore™-T200 instrument was used. Series S sensor chip Protein A (GE Healthcare) was applied to capture each antibody on different flow cells (FC) to achieve approximately 200 response units (RU), followed by the injection of five-fold serial dilutions of human or mouse MerTK (0.8 nM to 500 nM) in HBS-EP buffer (100 mM HEPES pH7.4, 150 mM NaCl, 3 mM EDTA, 0.05% (v/v) Surfactant P20) with a flow rate of 100 ul/min at 25°C. Association rates (kon) and dissociation rates (koff) were calculated using a simple one-to-one Langmuir binding model (BIAcore T200 evaluation software version 2.0). The equilibrium dissociation constant (KD) was calculated as the ratio koff/kon. The result is shown in Figure S4A.
Epitope binning
To bin all three MerTK agonistic antibody epitope, the microarray-based 96 × 96 microfluidic system (IBIS-MX96 SPRi, Carterra USA) was used. First, all three antibodies (10 ug/ml in 10 mM sodium acetate buffer pH4.5) were directly immobilized onto a sensorprism CMD 200M sensor chip (XanTec Bioanalytics, Germany) using amine-coupling chemistry in the instrument of continuous flow microspotting (CFM, Carterra, USA). Second, 250 nM human or mouse MerTK was injected over the sensor chip for 5 min binding, followed by another 4 min injection of each antibody (10 ug/mL in HBS-EP buffer) at 25°C. The chip surface was regenerated between each cycle using 10 mM glycine pH1.7, and the binding response was recorded and analyzed in Wasatch microfluidics’ binning software for network plotting. The result is shown in Figure S4B.
ELISA assay measuring antibody specificity
ELISA was carried out to determine the specificity of MerTK bispecific antibodies for other members of TAM family. Briefly, 1ug/ml of human MerTK (Genentech), human AXL (Creative Biomart, Catalog AXL-769H), human TYRO3 (Creative Biomart, Catalog TYRO3-195H), mouse MerTK (R&D systems, Catalog 591-MR-100), mouse AXL (Creative Biomart, Catalog AXL-626M), mouse TYRO3 (R&D systems, Catalog 759-DT-100) were coated onto ELISA plates. The plates were blocked with SuperBlock (TBS) blocking buffer (Thermo Fisher, Catalog 37535). hIgG1 and mIgG2a versions of bispecific antibodies were added followed by horseradish peroxidase (HRP)-conjugated anti-human IgG (Jackson laboratories, Catalog 709-035-149) and HRP-conjugated anti-mouse IgG (Jackson laboratories, Catalog 715-035-151) at 1:20,000 dilution. The plates were developed with TMB (Seracare, Catalog 5120–0047). The reaction was stopped with stop solution (Seracare, Catalog 5150–0020) and read at 450 nm using SpectraMax M5 (Molecular Devices). To test that both human and mouse AXL and TYRO3 were coated on the plate, binding was checked with anti-human AXL (Novus Biologicals, MAB154-500), anti-human TYRO3 (R&D systems, Catalog MAB859-500), anti-mouse AXL (R&D systems, Catalog MAB8541), and anti-mouse TYRO3 (R&D systems, Catalog BAM759). The result showed that all three antibody clones selectively bound MerTK, but not two other family members, AXL and TYRO3 (Figure S4C and D).
Immunoprecipitation and immunoblots
Antibodies for immunoblotting were pAKT (Cell signaling, Catalog 4060S), AKT (Cell signaling, Catalog 4691S), β-Actin (Abcam, Catalog ab8226), MerTK (Genentech, clone 13D8 and 14C9) and phosphotyrosine (EMD, Catalog 05–321). For immunoprecipitation, anti-MerTK antibody (Cell signaling, Catalog 4319S) was conjugated to anti-rabbit magnetic beads (Invitrogen, Catalog 11203D) and rotated overnight. M0 macrophages were serum starved for 2 hours. Dead/apoptotic cells from Raji cell culture were depleted using a dead cell removal kit (Miltenyi Biotec, Catalog 130-090-101). Cells with viability more than 95% (as determined by Vi-Cell) were used. Raji cells in serum-free media were incubated with anti-CD20/18G7 hIgG1-LALAPG at 5 ug/ml for 1 hour at room temperature. Raji cell-antibody mixture were added to macrophages and incubated at 37°C for 30 minutes. After, incubation cells were washed twice with phosphate-buffered saline (PBS) and macrophages were lysed through sonication with RIPA buffer (Thermo Fisher, Catalog 89901) supplemented with protease/phosphatase inhibitor cocktail (Thermo Fisher, Catalog 1861281). Lysates were combined with anti-MerTK–conjugated magnetic beads and rotated for 2 hours at 4°C. Beads were washed 3 times with RIPA buffer and immunoblotted.
Human primary macrophages
Human peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated from buffy coat using a ficoll gradient centrifugation. CD14+ monocytes were purified from PBMCs by negative depletion using a CD14 isolation kit without depletion of CD16+ cells (StemCell Technologies, Catalog 19058). About 20X10^6 cells were differentiated in 15cm2 plates to M0 for 7 days with 50 ng/ml macrophage colony stimulating factor (M-CSF, StemCell Technologies, Catalog 78057) in RPMI media containing penicillin/streptomycin, 10% heat inactivated fetal bovine serum (FBS), 2 mM glutamine (RPMI complete media). Additionally, a subset of M0 macrophages were polarized to M2 by adding 25 ng/ml of IL-10 (StemCell Technologies, Catalog 78024) on Day 4.
Primary bone marrow-derived macrophages (BMDM) and microglia culture
For BMDM culture, femurs from wild-type (WT) and MerTK−/- Citation23 were dissected and sterilized. Bone marrow was flushed with a syringe filled with RPMI complete media. The resulting marrow suspension was filtered through 100 μM nylon filter and treated with ACK lysis buffer to lyse red blood cells. Cells were washed twice with RPMI media and differentiated with 50 ng/ml M-CSF (StemCell Technologies, Catalog 78059) for 7 days in RPMI complete media. Additionally, a subset of M0 macrophages were polarized to M2 by adding 25 ng/ml of IL-10 (StemCell Technologies, Catalog 78079.1) on Day 4. For primary microglia culture, mixed glial cultures were harvested from whole brain from P2 mice and cultured in DMEM containing penicillin/streptomycin, 10% heat inactivated FBS, 2 mM glutamine. About 50% media was replaced every other day and on day 7, 10 ng/ml M-CSF was added. On Day 10–14, microglia were harvested by shaking the flask at 200 RPM (RCF 1g) for 1 hour (Rotamax 120). All mice were either generated at Genentech or obtained from Charles river laboratories and maintained in accordance with American Association of Laboratory Animal Care (AALAC) guidelines. The experiments were conducted in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Genentech Institutional Animal Care and Use Committee.
ELISA assay for measuring aβ binding
Aβ 1–42 (Anaspec, Catalog AS-24224) was treated with hexafluoroisopropanol (HFIP) and air dried overnight. HFIP-treated Aβ was dissolved in dimethyl sulfoxide (DMSO) by sonication. About 0.1 ug/ml DMSO treated Aβ in bicarbonate buffer was coated onto ELISA plates overnight. The plates were blocked with SuperBlock (TBS) blocking buffer (Thermo Fisher, Catalog 37535). For Aβ binding, mIgG2a versions of bispecific antibodies were added, followed by HRP-conjugated anti-mouse IgG (Jackson Laboratories, Catalog 715-035-151) at 1:20,000 dilution. The plates were developed with TMB (Seracare, Catalog 5120–0047). The reaction was stopped with stop solution (Seracare, Catalog 5150–0020) and read at 450 nm using SpectraMax M5 (Molecular Devices).
Homogeneous time-resolved fluorescence assay
Human primary macrophages (2X10^4) or mouse BMDMs (3X10^4) were plated in a 96-well plate. Cells were serum starved for 4 hours and stimulated with MerTK agonist antibodies for 10 mins. Phospho-AKT (Ser473) level was monitored using an HTRF assay (Cisbio, Catalog 64AKSPEH). For experiments with inhibitors, cells were pre-treated 30mins prior to stimulation with antibodies.
Phagocytosis of live Raji, live Jurkat or apoptotic Jurkat cells by macrophages
Nearly 1X10^5 M2 macrophages were seeded overnight in 24-well plate. Dead/apoptotic cells from the target (Raji and Jurkat) cell culture were depleted using a dead cell removal kit (Miltenyi Biotec, Catalog 130-090-101). Cells with viability more than 95% (as determined by Vi-Cell) were labeled with 0.5 μM carboxyfluorescein succinimidyl ester (CFSE), using a CFSE Cell Proliferation Kit (Life Technologies, Catalog C34554). About 5X10^5 labeled target cells in serum-free media were added to 1X10^5 macrophages (5:1) in the presence of indicated antibodies for 20 mins. After incubation, non-phagocytosed cells were washed with PBS. Macrophages were treated with TrypLE to prepare a cell suspension. Macrophages were incubated with Fc block for 15 mins and stained with CD14-APC (BioLegend Catalog 301808) and CD22-PEcy7(BioLegend Catalog 302514). Non-phagocytosed Raji cells that are bound to macrophages were removed from FACS analysis using a second marker for Raji cells (CD22-PEcy7).
For phagocytosis of apoptotic cells, CFSE labeled Jurkat cells were treated with staurosporine (2 μM) (Sigma, Catalog S6942) for 4 hours to induce apoptosis. About 5X10^5 CFSE labeled apoptotic Jurkat cells in RPMI complete media were added to 1X10^5 macrophages (5:1) in the presence of indicated antibodies for 20 mins.
The percentage of phagocytosis is defined as percentage of macrophages that are both CD14-APC+ and CFSE+. For experiments with inhibitors and blocking antibodies, cells were pre-treated with these agents 30 mins prior to carrying out the phagocytosis assay.
For ImageStream analysis, phagocytosis was carried out in a similar way as described above for flow cytometry analysis. Macrophages that had been stained were fixed in 4% paraformaldehyde. Cell suspension at concentration of 2X10^6/mL was prepared. Cell images were collected at ~ 50 cells per second using an IS100 ImageStream system (Amnis Corporation, Seattle, WA). Channel 2 was assigned as CFSE, channel 9 as bright field and channel 11 as allophycocyanin (APC). Data was processed with ImageStream Data Analysis and Exploration Software (IDEAS, Amnis). Percentage of phagocytosis is defined as percentage of macrophages that are both CD14-APC+ and CFSE+.
Phagocytosis of aggregated Aβ
Aβ 1–42 Hylite 488 (Anaspec, Catalog AS-60479-01) was treated with HFIP and air dried overnight. HFIP-treated Aβ was dissolved in DMSO by sonication. To promote aggregation, DMSO treated Aβ was diluted in PBS to a final concentration of 200 μM at 37°C for 3 days in a shaker. Aggregated Aβ was incubated with antibodies for 1 hour at 37°C. 3X10^4 BMDMs or primary microglia were plated in a 96-well plate. Aggregated Aβ-antibody complex were added to cells in serum free media for a final concentration of 1µM. After incubation, media was removed and non-phagocytosed Aβ was quenched with trypan blue (0.2% in PBS pH 4.4) for 1 min. Trypan blue was removed and fluorescence signals were measured at 485-nm excitation/538-nm emission. For experiments with inhibitors and blocking antibodies, cells were pre-treated with these agents 30 mins prior to carrying out phagocytosis assay.
Cytokine assays
Indicated antibodies at 10 ug/ml in PBS were coated onto 24-well tissue culture plates overnight at room temperature. After coating, plates were washed twice in PBS and 2X10^5 human primary macrophages (differentiated in M-CSF for 7 days) in 300ul of RPMI complete media was added. After 24 hours supernatant was collected for Luminex analysis (Millipore). TNF levels were measured with human (Abcam, Catalog ab181421) and mouse (R&D systems, Catalog MTA00B) ELISA kits.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author contributions
VK and MY designed the research and wrote the paper, with input from all the authors. VK performed all the experiments and analyzed the data. JB processed the Imagestream data. DE designed and purified bispecific antibodies. WL, YC and YW discovered MerTK antibodies.
Competing Interests
All authors are employees of Genentech, a member of the Roche group.
Supplemental Material
Download Zip (8.3 MB)Acknowledgments
We thank Andres Martinez for measuring cytokine levels by Luminex and Genentech FACS core for technical assistance.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Related Research Data
References
- Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–8.
- Morkuniene R, Zvirbliene A, Dalgediene I, Cizas P, Jankeviciute S, Baliutyte G, Jokubka R, Jankunec M, Valincius G, Borutaite V, et al. Antibodies bound to Abeta oligomers potentiate the neurotoxicity of Abeta by activating microglia. J Neurochem. 2013;126:604–15. doi:https://doi.org/10.1111/jnc.2013.126.issue-5.
- Bugelski PJ, Achuthanandam R, Capocasale RJ, Treacy G, Bouman-Thio E. Monoclonal antibody-induced cytokine-release syndrome. Expert Rev Clin Immunol. 2009;5:499–521. doi:https://doi.org/10.1586/eci.09.31.
- Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, Mathis CA, Blennow K, Barakos J, Okello AA, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363–72. doi:https://doi.org/10.1016/S1474-4422(10)70043-0.
- Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, van Dyck CH, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73:2061–70. doi:https://doi.org/10.1212/WNL.0b013e3181c67808.
- Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013;5:a009076. doi:https://doi.org/10.1101/cshperspect.a009076.
- Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196:135–40. doi:https://doi.org/10.1084/jem.20012094.
- Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–36. doi:https://doi.org/10.1016/j.cell.2007.10.034.
- Lee YJ, Han JY, Byun J, Park HJ, Park EM, Chong YH, Cho M-S, Kang JL. Inhibiting Mer receptor tyrosine kinase suppresses STAT1, SOCS1/3, and NF-kappaB activation and enhances inflammatory responses in lipopolysaccharide-induced acute lung injury. J Leukoc Biol. 2012;91:921–32. doi:https://doi.org/10.1189/jlb.0611289.
- Cai B, Kasikara C, Doran AC, Ramakrishnan R, Birge RB, Tabas I. MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci Signal. 2018;11(549).
- Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, Lohmann S, Piorkowska K, Gafner V, Atwal JK, et al. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci. 2012;32:9677–89. doi:https://doi.org/10.1523/JNEUROSCI.4742-11.2012.
- Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E, et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29:457–66. doi:https://doi.org/10.1093/protein/gzw040.
- Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes JM, Zhang Y, Lin YL, Chung S, Dennis MS, Zuchero YJY, et al. Effector-attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J Biol Chem. 2017;292:3900–08. doi:https://doi.org/10.1074/jbc.M116.767749.
- Atwell S, Ridgway JB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol. 1997;270:26–35. doi:https://doi.org/10.1006/jmbi.1997.1116.
- Sun LL, Ellerman D, Mathieu M, Hristopoulos M, Chen X, Li Y, Yan X, Clark R, Reyes A, Stefanich E, et al. Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies. Sci Transl Med. 2015;7:287ra70. doi:https://doi.org/10.1126/scitranslmed.aaa4802.
- Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol. 1989;143:2595–601.
- Dransfield I, Zagorska A, Lew ED, Michail K, Lemke G. Mer receptor tyrosine kinase mediates both tethering and phagocytosis of apoptotic cells. Cell Death Dis. 2015;6:e1646. doi:https://doi.org/10.1038/cddis.2015.18.
- Hezareh M, Hessell AJ, Jensen RC, van de Winkel JG, Parren PW. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J Virol. 2001;75:12161–68. doi:https://doi.org/10.1128/JVI.75.24.12161-12168.2001.
- Fuller JP, Stavenhagen JB, Christensen S, Kartberg F, Glennie MJ, Teeling JL. Comparing the efficacy and neuroinflammatory potential of three anti-abeta antibodies. Acta Neuropathol. 2015;130:699–711. doi:https://doi.org/10.1007/s00401-015-1484-2.
- Savage JC, Jay T, Goduni E, Quigley C, Mariani MM, Malm T, Ransohoff RM, Lamb BT, Landreth GE. Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer’s disease. J Neurosci. 2015;35:6532–43. doi:https://doi.org/10.1523/JNEUROSCI.4586-14.2015.
- Fourgeaud L, Traves PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, Callaway P, Zagórska A, Rothlin CV, Nimmerjahn A, et al. TAM receptors regulate multiple features of microglial physiology. Nature. 2016;532:240–44. doi:https://doi.org/10.1038/nature17630.
- Seeber S, Ros F, Thorey I, Tiefenthaler G, Kaluza K, Lifke V, Fischer JAA, Klostermann S, Endl J, Kopetzki E, et al. A robust high throughput platform to generate functional recombinant monoclonal antibodies using rabbit B cells from peripheral blood. PLoS One. 2014;9:e86184. doi:https://doi.org/10.1371/journal.pone.0086184.
- Anderson KR, Haeussler M, Watanabe C, Janakiraman V, Lund J, Modrusan Z, Stinson J, Bei Q, Buechler A, Yu C, et al. CRISPR off-target analysis in genetically engineered rats and mice. Nat Methods. 2018;15:512–14. doi:https://doi.org/10.1038/s41592-018-0011-5.