
ABSTRACT
Mucosal immunity is dominated by secretory IgA and IgM, although these are less favorable compared to IgG molecules for therapeutic development. Polymeric IgA and IgM are actively transported across the epithelial barrier via engagement of the polymeric Ig receptor (pIgR), but IgG molecules lack a lumen-targeted active transport mechanism, resulting in poor biodistribution of IgG therapeutics in mucosal tissues. In this work, we describe the discovery and characterization of single-domain antibodies (VHH) that engage pIgR and undergo transepithelial transport across the mucosal epithelium. The anti-pIgR VHH panel displayed a broad range of biophysical characteristics, epitope diversity, IgA competition profiles and transcytosis activity in cell and human primary lung tissue models. Making use of this diverse VHH panel, we studied the relationship between biophysical and functional properties of anti-pIgR binders targeting different domains and epitopes of pIgR. These VHH molecules will serve as excellent tools for studying pIgR-mediated transport of biologics and for delivering multispecific IgG antibodies into mucosal lumen, where they can target and neutralize mucosal antigens.
Introduction
The mucosal epithelial barrier is essential for life because it protects the body’s internal milieu from the external environment. It prevents the entry of pathogens and toxins into the body, while selectively allowing the movement of nutrients and proteins across mucosal epithelia through different mechanisms of transport. The mucosal barrier is composed of one or more layers of epithelial cells that secrete mucus, and an underlying layer of loose connective tissue termed lamina propria. Polarization of epithelial cells, featuring distinct apical and basolateral layers, is essential for bidirectional transport across the mucosal epithelial barrier.Citation1
A major driver of mucosal immunity is immunoglobulin (Ig) A. IgA is the most abundantly produced antibody in the human body, greater than all other immunoglobulin subtypes combined. Most IgA is produced as a dimer at mucosal sites, which harbor 80–90% of all Ig-producing plasma cells in the body. In serum, IgA is the second most abundant antibody subtype, after IgG. Serum IgA is produced by bone marrow-residing plasma cells and generally exists as a monomer.Citation2–Citation4 Only dimeric IgA (dIgA) is capable of binding to the polymeric Ig receptor (pIgR), which is responsible for transporting IgA across the mucosal epithelial barrier and for facilitating the conversion of dIgA into secretory IgA (sIgA).Citation5 Heavy chains of dimeric IgA feature an additional 18-residue C-terminal extension, termed the tailpiece, which interacts with a 137-residue polypeptide termed the joining chain. The joining chain is required by dimeric IgA to interact with pIgR.Citation6
Human pIgR (hpIgR) is an 82 kDa, single-pass transmembrane receptor containing a 620-residue extracellular domain (ECD), a 23-residue transmembrane domain and a 103-residue intracellular domain.Citation7 hpIgR ECD consists of five Ig domains (domain-1 to domain-5, residues 1–545) that take on a compact, globular structure in the absence of bound IgA (Figure S1A). In this compact form, domain-1 is situated between domain-2 and domain-5 such that residues that are important for interaction with dIgA are involved in these intramolecular interactions.Citation8 Solution x-ray scattering studies suggest that upon interaction with dIgA, pIgR adopts an extended conformation, with domain-1 interacting with the Cα2 domain of one Fcα subunit and domain-5 binding the Cα2 subunit on the same side of the opposite Fcα subunit (Figure S1B).Citation9 This asymmetric binding of pIgR across both subunits of Fcα explains its inability to effectively bind to monomeric IgA, and its interactions with the joining chain support the 1:2 stoichiometry of binding.
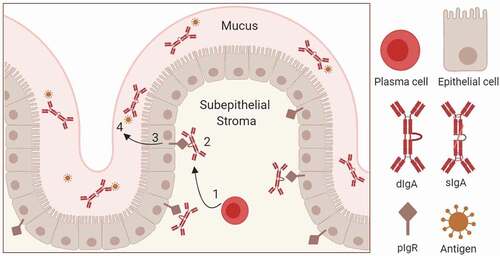
pIgR transcytosis pathway starts with synthesis of pIgR and dIgA. pIgR is synthesized by secretory epithelial cells and delivered to the basolateral membrane of the epithelium. dIgA, produced by plasma cells in the lamina propria, first interacts with the joining chain, and subsequently with pIgR ECD.Citation7,Citation10 The pIgR-dIgA complex is endocytosed and transcytosed to the apical layer by vesicular transport, during which a disulfide bond can be formed between hpIgR ECD (C467 of domain-5) and dIgA (C311 of Cα2 domain). Upon reaching the apical surface, hpIgR ECD gets cleaved at the C-terminal tail (residue 585) and released into the mucosal lumen along with dIgA. The portion of hpIgR ECD (residues 1–547) that remains to be bound with dIgA is known as secretory component and complex between dIgA and secretory component is known as sIgA.Citation11,Citation12 In the mucosal lumen, the secretory component has numerous functions, including conferring stability and mucophilic properties to dIgA, nonspecific microbial scavenging, neutralization of toxins and epithelial homeostasis.Citation13 The schematic of pIgR-mediated transport of dIgA is shown in .
Figure 1. Schematic of pIgR-mediated dimeric IgA transport across the mucosal epithelial barrier. (1) IgA Production by plasma cells and IgA dimerization; (2) binding of dimeric IgA (dIgA) to pIgR ECD on the basolateral side of the epithelium. pIgR-dIgA interactions are mediated by domains 1 and 5 of pIgR and Fc and J chains of dIgA; (3) pIgR-mediated transcytosis of dimeric IgA. Clathrin-mediated endocytosis drives the basolateral to apical transport. Upon reaching the apical side, pIgR ECD is proteolytically cleaved and released into mucus along with IgA. Mucosal IgA in complex with secreted pIgR ECD (secretory component) is termed as secretory IgA (sIgA); and (4) Neutralization of mucosal antigens by sIgA
Of the five Ig subtypes, only polymeric IgA and IgM can cross the epithelial barrier and reach the mucosal lumen by engaging with pIgR.Citation7,Citation11,Citation13 Though there are some examples of using IgA as potential therapeutics,Citation14 IgG-based molecules are overwhelmingly favored for therapeutic antibody development due to their relative ease of production, long serum half-lives, and well-characterized effector functions. However, IgG molecules cannot be secreted into mucosa and rely only on passive transport to cross the epithelial barrier. This results in very poor accumulation of IgG molecules in mucosa upon intravenous administration, which is undesirable for therapeutic IgG antibodies targeting mucosal organs (e.g., lungs, intestine) for treating respiratory, pulmonary and intestinal diseases.Citation15 Previous studies have shown that conferring pIgR-binding abilities to IgG molecules and other biologic payloads can result in the transcytosis of such molecules across the epithelial barrier. Other groups have generated pIgR-binding peptides,Citation15–Citation17 IgG antibodies, antigen-binding fragments (Fabs), single-chain variable fragments,Citation18–Citation24 and single-domain antibodies (VHH)Citation25 to capitalize on the transcytosis activity of pIgR. However, no binders have been reported against the dIgA-binding domain-1 of pIgR. Also, knowledge of the biophysical and functional properties of binders targeting different domains and epitopes of pIgR is still lacking in the field.
To overcome the mucosal epithelial barrier by exploiting pIgR-mediated active transport, we discovered and characterized a panel of anti-hpIgR VHHs. The anti-pIgR VHH panel displayed a broad range of biophysical characteristics, epitope diversity, dIgA competition profiles and transcytosis activity in epithelial monolayers. In a primary lung tissue model, anti-pIgR VHHs could function as a “trojan horse” for delivering biological payloads into the apical mucus from the basolateral epithelium. For the first time, we report the discovery of binders against domain-1 and domain-2 of pIgR ECD and show that high-affinity interaction on dIgA-binding domain-1 alone is not sufficient for transcytosis. Two VHHs with overlapping epitopes on pIgR domain-1 showed opposing results in transcytosis assays. Domain-2 binding VHHs showed relatively lower pIgR staining than other VHHs in tissue models post-transcytosis, suggesting that they may increase pIgR secretion. VHHs from this work will be useful for characterizing pIgR-mediated transport and for delivering therapeutic IgG antibodies and other payloads into the mucosa.
Results
Discovery and biophysical characterization of pIgR binders
To generate antibodies targeted against pIgR, we immunized llamas with recombinant hpIgR and mouse pIgR (mpIgR) ECD constructs, isolated B lymphocytes that were positive for VHH and antigen binding, and recovered, cloned and sequenced VHH genes using established protocols. Following VHH-region sequencing, we expressed and purified a panel of 73 VHH molecules as fusions to the human IgG1 mono-Fc protein (mFc).Citation26 Screening this VHH panel for binding to hpIgR and mpIgR ECD by ELISA resulted in 40 positive hits. Bio-layer interferometry showed that 14 VHHs from this panel had KD lower than 100 nM for binding to the mpIgR or hpIgR ECD. Five VHHs were selective for mpIgR, six VHHs were selective for hpIgR and three VHHs cross-reacted with both mpIgR and hpIgR (data not shown). We selected 10 hpIgR-binding VHHs from this panel for further characterization: two cross-reactive VHH with KD <100 nM for mpIgR and hpIgR, six hpIgR-selective VHH with KD <100 nM and two hpIgR-selective VHH with KD >100 nM ().
Table 1. Tm, KD and EC50 values of anti-pIgR VHHs
We purified these 10 VHH-mFc molecules from ExpiCHO cells by Protein-A affinity chromatography and assessed the quality of purified molecules by analytical-size exclusion chromatography (A-SEC) and size exclusion chromatography combined with multi-angle light scattering (SEC-MALS). Purification yield, % monomer content and molecular weight of VHH-mFc molecules are listed in Table S1. A-SEC showed that % monomer content ranged from 80% to 99%. SEC-MALS showed that molecular weight of VHH-mFc molecules ranged from 41.3 kDa to 48.7 kDa, consistent with sequence-based prediction. Next, we measured thermal stability and binding kinetics of VHH-mFc molecules by differential scanning fluorimetry and bio-layer interferometry, respectively. Results from these assays are summarized in . Tm values of 10 VHH-mFc molecules (Tm corresponding to VHH unfolding) ranged from 53.9°C to 76.4°C. Except for VHH7 and VHH10, KD values of pIgR VHHs ranged from 4 nM to 34 nM. To test binding of VHHs to cell-surface hpIgR, we used Madin-Darby canine kidney (MDCK) cells engineered to stably express full-length hpIgR. Flow cytometry showed that six VHHs had EC50 values of <10 nM for binding to MDCK-hpIgR cells. The EC50 of four other VHHs ranged from 12 nM to 37 nM ().
Functional characterization of pIgR binders
We used hpIgR expressing MDCK cells, a commonly used epithelial model systemCitation27 to test the transcytosis activity of VHH-mFc molecules. Expression of hpIgR in MDCK cells and monolayer formation were confirmed by confocal laser microscopy using a commercial anti-pIgR antibody (Figure S2). To test the transcytosis activity of VHH-mFc molecules across MDCK-hpIgR monolayers, 20 μg of test or control VHH-mFc molecules were added to the basolateral chamber and the amount of VHH-mFc present in the apical chamber was quantified at 0, 24 and 48 hours by electrochemiluminescence method (Figure S3). We used a biotinylated anti-VHH antibody to capture VHH-mFc on streptavidin plates and a ruthenylated anti-Fc antibody to detect VHH-mFc by the Meso Scale Discovery (MSD) platform. Six VHHs (2, 4, 6, 9, 11 and 12) showed >10-fold increase in their apical concentration relative to control VHH ().
Figure 2. Functional validation of VHH-mFc molecules. (a) Transcytosis activity of VHH-mFc molecules across MDCK-hpIgR monolayers from the basolateral to the apical chamber. Fold increase in apical VHH amount at 24 hours relative to control VHH is shown in 2A. Apical VHH at 0, 24, and 48 hours is shown in Figure S3. (b) Transcytosis activity of VHH-mFc molecules in the EpiAirway primary human lung tissue model. Fold increase in mucosal VHH amount at 24 hours relative to control VHH is shown in 2B. Mucosal VHH at 0, 24, and 48 hours is shown in Figure S4. (c) Photomicrographs of fixed, permeabilized, and hpIgR & VHH stained cross-sections closer to the apical epithelium (2 days post-treatment). Immunofluorescence staining: nucleus (blue), hpIgR (green) and VHH (red). VHH14 was used as a negative control
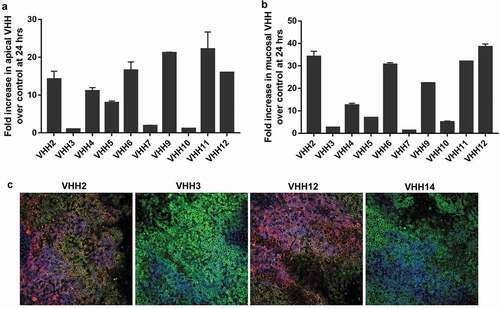
To test whether VHH-mFc molecules can cross the epithelial barrier and get secreted into mucus, we used the EpiAirway human lung tissue model. The EpiAirway model, engineered from primary human bronchial epithelial cells, is a fully differentiated tissue model that mimics in vivo-like structure and function.Citation28,Citation29 To measure the transcytosis activity of VHH-mFc molecules, we added 20 μg of VHH-mFc to the basolateral chamber and quantified the amount of VHH-mFc present in the apical mucus at 0, 24 and 48 hours post treatment by electrochemiluminescence method (Figure S4). Five VHHs (2, 6, 9, 11 and 12) showed >20-fold increase in their mucosal amount relative to control VHH molecules (). VHH12 showed 38-fold increase in mucus relative to control VHH and displayed the highest transcytosis activity.
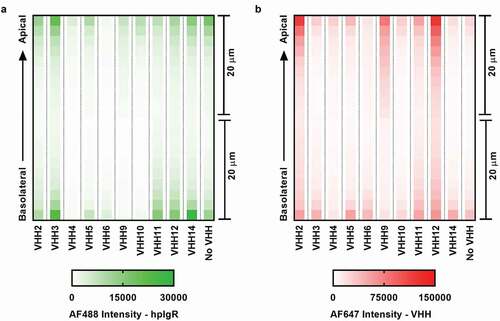
Next, we fixed, permeabilized and stained EpiAirway tissues, and used indirect immunofluorescence to visualize hpIgR and VHH closer to the apical epithelium (48 hours post-treatment). Photomicrographs of tissue cross-sections for four VHH samples (2, 3, 12, and 14) are shown in Figure S5. Merged images are shown in . Consistent with transcytosis results, VHH2 and VHH12-treated tissue samples stained strongly for VHH and colocalized with pIgR relative to VHH3 and VHH14 (negative control). To trace the amount and location of hpIgR and VHH across the tissue section, we imaged the EpiAirway model along the Z-axis by confocal laser microscopy (). VHHs displayed distinct profiles of pIgR and VHH distribution across the tissue depth dimension. Among the five VHHs that showed potent transcytosis, VHH2, VHH9 and VHH12-treated tissue models showed higher VHH staining near the apical surface than the other VHHs. Interestingly, VHH6-treated model showed the lowest staining for both VHH and pIgR across the tissue thickness.
Figure 3. Tracking pIgR and VHH across the primary human lung tissue model. (a): Heatmap showing the amount of pIgR retained in the tissue model following transcytosis. (b) Heatmap showing the amount of VHH retained in the tissue model following transcytosis. Following 48 hours post-VHH treatment, tissue samples were fixed, permeabilized and stained for hpIgR and VHH. A mouse anti-hpIgR primary antibody and Alexa-Flour 488-labeled anti-mouse secondary antibody were used to stain hpIgR. A biotinylated anti-VHH primary antibody and Alexa-Flour 647-labeled streptavidin were used to stain VHH. Indirect immunofluorescence images were collected and processed using Opera Phenix confocal laser microscopy. VHH14 was used as a negative control
Epitope binning and VHH/IgA competition studies
To conduct domain-level epitope mapping of VHHs, we successfully expressed and purified seven HIS-tagged hpIgR domain constructs (denoted D1, D2, D3, D5, D1-D2, D2-D3 and D4-D5) each encoding one or two domains of hpIgR ECD from HEK293 cells using immobilized metal ion affinity chromatography. Two constructs, D4 and D3-D4, showed poor expression and purification yields, and therefore they were not used for epitope mapping assays. We tested the binding of VHH-mFc molecules to immobilized pIgR constructs by the electrochemiluminescence method. Results from the binding assay are shown as a heat map in . In brief, the epitopes of VHH2 and VHH3 are primarily contained within hpIgR domain-1, and the epitopes of VHH4 and VHH6 are primarily contained within hpIgR domain-2. The epitopes of other six VHHs are primarily contained within hpIgR domains 4–5.
Figure 4. Epitope binning and binding competition between VHH and dIgA2. (a) Domain-level epitope mapping of pIgR binders. The heat map shows binding of VHH-mFc molecules to immobilized pIgR constructs in electrochemiluminescence units. VHH1 was used as a negative control. (b) The effect of dIgA2 on hpIgR-VHH binding. KD for full-length hpIgR ECD binding to immobilized VHH-mFc in the absence (blue) and presence (red) of dIgA2. (c) The effect of VHH on hpIgR-dIgA2 binding. KD for full-length hpIgR ECD binding to dIgA2 in the absence (blue) and presence (red) of VHH-mFc molecules
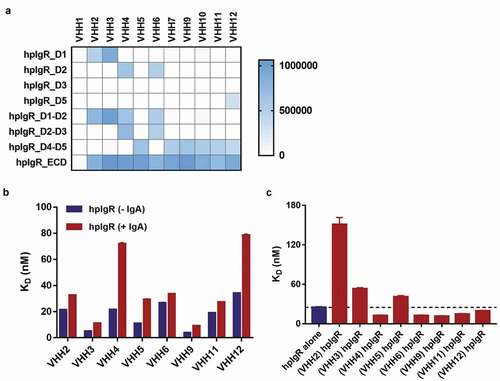
Next, we conducted competition binding assays for eight VHH-mFc molecules that displayed KD values of <100 nM for binding to hpIgR. First, to test the influence of IgA on hpIgR-VHH binding, we measured KD values for full-length hpIgR ECD binding to immobilized VHH-mFc molecules in the absence and presence of dIgA2 by bio-layer interferometry (). In total, VHHs showed a 1.3- to 3.3-fold decrease in affinity for binding to hpIgR ECD due to the presence of dIgA. Pre-bound IgA had a small negative effect on binding of VHHs to pIgR, possibly due to steric hindrance arising from bound dIgA or conformational rearrangement of hpIgR ECD. Second, to test the influence of VHH on hpIgR-IgA binding, we measured KD values for full-length hpIgR ECD binding to immobilized dIgA2 in the absence and presence of VHH-mFc molecules by bio-layer interferometry (). VHH2 had a negative effect on IgA binding to pIgR. VHH3 and VHH5 displayed a small negative effect on IgA binding to pIgR. Given that hpIgR domain-1 is necessary for dimeric IgA binding,Citation30,Citation31 both domain-1 binders (VHH2 and VHH3) had a negative effect on IgA binding to hpIgR and showed direct competition with IgA. Interestingly, four VHHs (4, 6, 9 and 11) showed a small positive effect on IgA binding to pIgR.
Differential recognition of pIgR domain-1 by VHH2 and VHH3
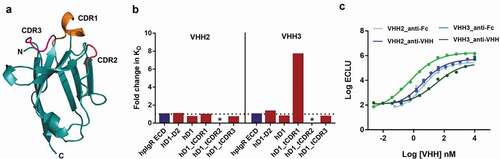
Epitopes of both human/mouse cross-reactive binders (VHH2 and VHH3) were primarily contained within pIgR domain-1. Among the two domain-1 binders, VHH2 showed high transcytosis activity, whereas VHH3 tested negative for transcytosis in both cell and tissue models, therefore we sought to test differences in VHH2 and VHH3 binding to hpIgR domain-1. First, to test the importance of hpIgR domain-1 complementary-determining regions (CDRs) on VHH2 and VHH3 binding, we swapped each domain-1 CDR of human pIgR () with the respective domain-1 CDR of teleost fish pIgR and purified three new CDR-swapped hpIgR domain-1 constructs by immobilized metal-ion chromatography. We selected pIgR domain-1 CDRs from teleost fish for CDR swaps because pIgR first emerged in teleost fish during evolution,Citation32 and thus we expected these mutations would increase the evolutionary distance of hpIgR domain-1 while maintaining proper folding of the domain-1 Ig fold. Also, the structure of teleost fish pIgR has been solved, and hpIgR domain-1 proteins harboring teleost fish CDRs have previously been validated.Citation8 We measured KD values for VHH2 and VHH3 binding to wild-type (WT) D1, WT D1-D2 and CDR-swapped hpIgR D1 constructs by bio-layer interferometry (Figure S6A) and calculated fold-change in KD values relative to WT full-length hpIgR ECD (). VHH2 and VHH3 showed similar binding profiles toward CDR2 and CDR3 of hpIgR domain-1, whereas they showed different binding profiles toward CDR1 of hpIgR domain-1. This indicated that VHH2 and VHH3 overlap partial epitopes on domain-1. The decrease in binding of VHH3 to hpIgR-D1_tCDR1 relative to hpIgR-D1 was mainly due to dissociation kinetics (increase in Koff) (Figure S6B and S6C).
Figure 5. Differential recognition of hpIgR domain-1 by VHH2 and VHH3. (a) Cartoon representation of hpIgR domain-1 created from PDB ID 5D4K. CDR1, CDR2 and CDR3 of hpIgR domain-1 are shown in orange, pink and light red, respectively. hpIgR domain-1 CDRs were swapped with corresponding teleost fish CDRs to test the influence of hpIgR domain-1 CDRs on VHH binding. (b) Fold change in KD for VHH2 and VHH3 binding to pIgR domain constructs relative to full-length hpIgR ECD. Kinetic parameters for VHH2 and VHH3 binding to pIgR protein constructs are shown in Figure S6. (c) Binding of VHH-mFc to immobilized full-length hpIgR ECD detected using either an anti-Fc secondary antibody or an anti-VHH secondary antibody
Second, to characterize differences in binding between VHH2 and VHH3, we measured their EC50 for binding to full-length hpIgR ECD protein by electrochemiluminescence method using two different detection antibodies, an anti-Fc antibody and an anti-VHH antibody. The increase in EC50 due to anti-VHH detection was used as a measure to determine differences in binding between VHH2 and VHH3. While no differences in EC50 were observed for VHH2 (4 nM for both detection antibodies), VHH3 showed a 54-fold increase in EC50 due to anti-VHH detection (). The epitope for the anti-VHH detection antibody was not readily available on the VHH3-pIgR complex relative to the VHH2-pIgR complex. This epitope masking could have occurred for many reasons, such as the anti-VHH epitope was hidden in the pIgR-VHH3 complex, and differences in angle of contact between VHH2 and VHH3. Taken together, these experiments indicated that VHH2 and VHH3 recognize domain-1 in a different fashion, which could have contributed to their differences in function.
Discussion
Of the five antibody subtypes, polymeric IgA and IgM overcome the mucosal epithelial barrier by pIgR-mediated active transport, but IgA and IgM are not favored for therapeutic antibody development. Therapeutic IgG antibodies do not bind pIgR and lack a lumen-targeted active transport mechanism, and therefore show very poor accumulation in mucosa upon intravenous administration. Previous studies have demonstrated that conferring pIgR-binding abilities to IgG and other biologic payloads can result in the transport of such molecules across the epithelial barrier.Citation15–Citation25 Thus, we sought to generate VHHs that could be transported via pIgR for use as a delivery moiety in multi-specific antibodies.
Even though pIgR-binding peptides, IgGs, Fabs, scFvs and VHHs have been generated previously, knowledge of the biophysical and functional properties of pIgR binders targeting different domains and epitopes of pIgR is still lacking in the field. While two antibodies binding to pIgR domain-5 and three antibodies binding to both domain-5 and C-terminal tail have been reported,Citation24 no binders have been reported against ligand-binding domain-1 and domain-2. One anti-pIgR peptide has been shown to compete with pneumococcal surface protein C that binds to pIgR between domains 3 and 4.Citation17 While this peptide and antibodies above did not compete with IgA,Citation17,Citation24 all four reported VHHs competed with IgA, but the epitopes of anti-pIgR VHHs are not known.Citation25 Surface plasmon resonance-based and cell-based binding constants were reported for antibodies and VHHs, respectively.Citation17,Citation25 Regarding transcytosis activity, most of the pIgR binders were validated only by cell-based assays. Three publications have shown in vivo activity for pIgR binders in mouse models.Citation15,Citation19,Citation23 In the most recent work, an anti-P. aeruginosa IgG fused with pIgR-binding peptides exhibited enhanced localization to the bronchoalveolar space, maintained Fc-mediated functional activity and promoted enhanced survival in an acute pneumonia model.Citation15
Given that the VHH scaffold is small (12–15 kDa) and well-recognized for high stability, solubility, cleft recognition properties, low production costs and easy genetic cross-linking,Citation33,Citation34 we generated VHHs against pIgR to develop a mucosal drug delivery platform. Following llama immunization, B-cell sorting and VHH gene isolation, we expressed, purified and screened a panel of 73 VHH-mFc molecules for binding to mpIgR and hpIgR ECD. The mono-Fc fusion served as a payload without affecting the valency of VHH. The 10 VHHs we selected for further characterization possessed KD values from ~5–500 nM for binding to hpIgR ECD and EC50 values from ~2–36 nM for binding to cell-surface hpIgR. Epitopes of these 10 VHHs were primarily contained within domain-1, domain-2 and domains 4–5 of hpIgR. To our knowledge, this is the first report showing the discovery of binders against domain-1 and domain-2 of pIgR ECD.
In this work, we used both engineered cell and primary human lung tissue models to test the transcytosis activity of VHHs. Five VHH molecules (2, 6, 9, 11 and 12) showed potent transcytosis activity in both cell and tissue models. Of the five VHHs, VHH9 had the lowest KD (4 nM) for binding to hpIgR ECD. The KD for four other VHHs ranged from 19 nM to 34 nM. For the five potent VHHs, the EC50 for binding to cell-surface hpIgR ranged from 2 nM to 12 nM, and Tm values ranged from 61°C to 70°C. Epitopes of these VHHs spanned different pIgR domains: domain-1 for VHH2, domain-2 for VHH6 and domains 4–5 for three other VHHs. Interestingly, binding to pIgR or specific domain(s) on pIgR is neither necessary nor sufficient for transcytosis. Despite binding to the same pIgR domain and having a 4-fold lower KD than VHH2, VHH3 showed much weaker transcytosis activity than VHH2 (2.6-fold vs 34.2-fold increase in mucus). Similarly, VHH5 displayed a 3-fold lower KD than VHH12 and both VHHs bound to pIgR domains 4–5, but VHH5 showed weaker transcytosis than VHH12 (6.9-fold vs 38.6-fold increase in mucus). These findings suggest that inducing a right conformational change in pIgR may be needed for transcytosis of pIgR binders. In our opinion, characterization of the binding-induced conformational change in pIgR and its role in transcytosis are critical factors for biologics utilizing the pIgR delivery platform.
We used confocal laser microscopy to track the location and amount of pIgR and VHH retained in the primary human lung tissue model post transcytosis. We observed distinct profiles of pIgR and VHH distribution across the depth dimension of the tissue model for different VHHs. Some VHHs that showed potent activity in the transcytosis assay also demonstrated high VHH distribution at the apical surfaces (such as VHH 2, 9, 12), while others (such as VHH6) performed well in the transcytosis assay but showed much less apical VHH staining. Interestingly, VHH6 also shows much less pIgR staining throughout the tissue thickness. Considering both pIgR domain-2 binders (VHH4 and VHH6) showed relatively less pIgR staining and relatively high mucus-related matrix effects during MSD assays, we speculate that these domain-2 binders induce pIgR secretion, possibly by conformational rearrangement of pIgR ECD. Further studies are required to understand the recycling kinetics of pIgR and VHHs during transcytosis experiments.
Consistent with previous findings,Citation17,Citation24 we show that binding competition with IgA is dispensable for pIgR-mediated transcytosis. Only VHH2 showed potent transcytosis and inhibited the binding of IgA to hpIgR ECD. Four other VHHs that showed potent transcytosis (VHH 6, 9, 11 and 12) did not demonstrate direct competition with IgA. These VHHs could be used for delivering payloads to mucosal tissues without interfering regular IgA trafficking and potentially exploting the same pIgR-mediated transport mechanism used by dIgA. Given this information, VHH12 would make a better delivery agent than VHH2 because it shows similar transcytosis activity but does not compete with normal IgA-pIgR interaction.
Domain-1 of pIgR ECD is crucial for interaction with dIgA.Citation30,Citation31 Two hpIgR domain-1-binding VHHs resulted from this work (VHH2 and VHH3) and both were cross reactive with mpIgR. While VHH2 showed potent transcytosis, VHH3 displayed very poor transcytosis activity in both cell and tissue models. Binding assays showed that VHH2 and VHH3 target partially overlapped epitopes on hpIgR domain-1 and recognize domain-1 in a different fashion. Interestingly, imaging studies showed that VHH3 retained more pIgR in the lung tissue model than other VHHs. Given that the domain-1 plays a crucial interface and role in the inactive to active transitioning of hpIgR,Citation8 we speculate that VHH3 binding could shift the pIgR equilibrium toward an inactive conformation. It will be interesting to test whether VHH3 is an antagonist of pIgR transcytosis.
While the primary function of pIgR is to transport polymeric IgA and IgM from the basolateral epithelium to the apical lumen (forward transcytosis), recycling of unliganded and uncleaved pIgR allows transport in the apical to the basolateral direction (reverse transcytosis).Citation35 This retrograde transport is exploited by S. pneumoniae to invade the epithelium and enter the circulation.Citation36 Gupta et al., showed that IgGs binding to domain-5 of pIgR ECD showed forward transcytosis, whereas IgGs binding to domain-5 and C-terminal tail of pIgR ECD showed reverse transcytosis.Citation24 It will be interesting to test whether domain-5 binding VHHs from this work can undergo reverse transcytosis.
In summary, we discovered VHHs binding to different pIgR domains and characterized them by biophysical and functional assays. Anti-pIgR VHHs showed varying degrees of biophysical characteristics, epitope diversity, IgA competition profiles and transcytosis activity in engineered cell and primary human lung tissue models. Making use of this diverse VHH panel, we studied the relationship between biophysical and functional properties of anti-pIgR binders targeting different domains and epitopes of pIgR. VHHs from this work will serve as excellent tools for characterizing pIgR-mediated transport and as “trojan horse” for delivering therapeutic IgG antibodies and other payloads into the mucosa.
Materials and methods
Generation and screening of anti-pIgR antibodies
To generate a panel of anti-pIgR single-domain antibodies, we immunized llamas with recombinant hpIgR and mpIgR ECDs (R&D systems) for ~90 days. Peripheral blood mononuclear cells were isolated from whole blood and B cells positive for VHH and antigen binding were sorted using a fluorescence-activated cell sorter. RNA and subsequently cDNA were prepared from sorted cells using standard kits. After first-strand cDNA synthesis, llama-specific primers annealing to the VH (heavy-chain variable region) and VHH leader sequence genes and to the CH2 gene were used to PCR amplify the VH and VHH gene repertoires. VHH repertoires were separated from VH repertoires by running the PCR fragments on a gel and excising the smaller band. The VHH gene repertoire was reamplified and cloned into a CMV-based mammalian vector in the VHH-hinge-human IgG1 mono-Fc format. ~200 unique sequences were identified by sequencing individual clones from the VHH library. 39 VHH sequences were chosen from the mpIgR campaign and 35 VHH sequences were chosen from the hpIgR campaign for small-scale expression and purification. Clone selection was based on sequence uniqueness (weighted heavily on CDR3) and a framework 2 signature indicative of VHH derived sequences.
Expression and purification of VHH-mFc molecules
Expression plasmids encoding VHH-mFc molecules were transfected into ExpiCHOTM cells according to manufacturer’s instructions. Cell supernatants were harvested after 6–7 days by centrifugation (4,000 g, 15 min), passed through a 0.45-μm filter, and purified by MabSelectTM SuReTM chromatography on an ÄKTA express system using phosphate-buffered saline (PBS) as running buffer and 0.1 M sodium acetate, pH 3.5 as elution buffer. Eluted fractions were immediately neutralized using 25% (v/v) 2 M Tris-HCl pH 7.0, dialyzed to PBS, sterilized by 0.22-μm filtration and stored at 4°C. Protein concentration was determined by UV-visible spectroscopy.
Quality assessment of purified VHH-mFc molecules
All purified VHH-mFc molecules were analyzed by analytical high-pressure liquid chromatography on an Agilent 1200 infinity system using an Agilent AdvanceBio Size exclusion column (300 Å, 2.7 μm, 4.6 × 150 mm). Column was equilibrated with 0.2 M sodium phosphate pH 6.8 and 20 μL of samples were injected at a concentration of 0.5 mg/ml and at a flow rate of 0.35 mL/min. Monomeric VHH-mFc elutes were detected at the expected retention time of ~4 min at these settings. Data analysis was performed in OpenLab Chemstation to calculate % monomer content. Molecular weight for purified VHH-mFc molecules was measured by size-exclusion chromatography combined with multi-angle light scattering. The experiment was performed on a Waters high-pressure liquid chromatography instrument connected in series to Wyatt μDAWN light scattering/μTrEX instrument. An Acquity UPLC Protein BEH size-exclusion column (200Å, 1.7 µm, 4.6 × 150 mm) was equilibrated with PBS pH 7.4 and 10 μL of samples were injected at a concentration of 0.5 mg/ml and at a flow rate of 0.3 mL/min. Molecular weight of the primary species (monomeric VHH-Fc) was calculated using the Astra software package (Wyatt).
Thermal stability of VHH-mFc molecules
Thermal stability of VHH-mFc molecules was determined by Nano-differential scanning fluorimetry using an automated Prometheus NT_Plex instrument (Nanotemper). Samples were diluted in PBS (pH 7.4) at 0.5 mg/mL concentration and loaded into 24-well capillaries in duplicates. A linear temperature ramp was applied from 20°C to 95°C at a rate of 1.0°C/minute. Intrinsic tryptophan and tyrosine fluorescence were monitored at the emission wavelengths of 330 nm and 350 nm and this ratio (F350 nm/F330 nm) was plotted against temperature to generate an unfolding curve. Data were analyzed and thermal unfolding parameters were calculated using the PR_ThermControl software (Nanotemper).
Dimeric IgA production
Recombinant human dimeric IgA was cloned, expressed and purified from Chinese hamster ovary (CHO) cells. Heavy, light and J-chains of IgA were cloned into mammalian expression vectors using the In-Fusion® HD Cloning Kit and co-transfected into ExpiCHOTM cells according to manufacturer’s instructions. Cell supernatants were harvested after 6–7 days by centrifugation, passed through a 0.45-μm filter, and purified by HiTrap LambdaFabSelectTM chromatography followed by size exclusion chromatography on an ÄKTA express system. Eluted fractions were dialyzed to PBS, sterilized by 0.22-μm filtration and stored at 4°C. Protein concentration was determined by UV-visible spectroscopy.
Bio-layer interferometry
Binding kinetic studies were conducted using the ForteBio Octet RED384 system (Pall Corporation). To measure binding kinetics between VHH-mFc molecules and HIS-tagged pIgR proteins (full-length ECD or individual domains), VHH-mFc was immobilized on amine-reactive generation-2 (ARG2) biosensors according to manufacturer’s instructions and increasing concentrations of pIgR proteins were exposed to sensor-immobilized VHH. In some cases, HIS-tagged pIgR proteins were immobilized on anti-HIS biosensors and exposed to increasing concentrations of VHH-mFc molecules. Association and dissociation rates were measured by the shift in wavelength (nm). For each sensor-immobilized protein, at least three different ligand concentrations were used, and KD (equilibrium dissociation constant) was obtained by fitting the data to 1:1 binding model. All reactions were performed at 25°C in PBS. Data were collected with Octet Data Acquisition version 7.1.0.87 (ForteBio) and analyzed using Octet Data Analysis version 7.1 (ForteBio).
To test the effect of IgA on pIgR-VHH binding, we measured KD values for pIgR ECD binding to VHH in presence of IgA. VHH immobilized on ARG2 biosensors was exposed to increasing concentrations of pIgR-IgA complex. KD values were obtained by fitting the data to 1:1 binding model. To measure binding kinetics between IgA and pIgR proteins, IgA was immobilized on ARG2 biosensors according to manufacturer’s instructions, and immobilized IgA was exposed to increasing concentrations of pIgR ECD. To test the effect of VHH on pIgR-IgA binding, we measured KD values for pIgR ECD binding to IgA in the presence of VHH. IgA immobilized on ARG2 biosensors was exposed to increasing concentrations of pIgR-VHH complex, and association and dissociation rates were measured by the shift in wavelength (nm). KD values were obtained by fitting the data to 2:1 binding model.
Flow cytometry
To test whether VHH-mFc molecules recognize cell-surface hpIgR, we used MDCK cells that stably express full-length hpIgR (engineered in-house). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) at 37°C with 5% CO2. Cells were split into equal fractions (≈70,000 cells) and incubated with increasing concentrations of VHH-mFc molecules for 30 min at 4°C. Cells were washed twice with cold PBS (pH 7.4) and incubated with a fluorescently-labeled anti-Fc antibody (Jackson Immuno-Research 109-606-170) for 30 min in staining buffer at 2 μg/ml at 4°C. Cells were washed twice with cold staining buffer, resuspended in running buffer and analyzed with an iQue Screener (Intellicyt Corporation). Binding was assessed by RL1 (A647) Geomeans from the live cell population and EC50 was calculated by fitting log VHH concentration versus MFI in Prism (GraphPad).
Transcytosis activity across MDCK-hpIgR monolayers
We used hpIgR-expressing MDCK cells, a commonly used epithelial model system to test the transcytosis activity of VHH-mFc molecules. Cells were cultured in DMEM containing 10% FBS at 37°C with 5% CO2. To prepare MDCK monolayers, 5 × 105 cells were seeded on fibronectin- and collagen-treated TranswellTM permeable supports (Costar) containing 0.4 μm polyester membrane filter. The cells were incubated for 3 days, serum starved for 2 hours and supplemented with DMEM containing 1% FBS (assay media). Basolateral and apical chambers contained 1.5 ml and 0.5 ml of assay media, respectively. To test the transcytosis activity of VHH-mFc molecules across MDCK-hpIgR monolayers, 20 μg of test or control VHH-mFc molecules were added to the basolateral chamber and 100 μL of media was collected from the basolateral and apical chambers at 0, 24 and 48 hours. The amount of VHH present in basolateral and apical media was quantified by electrochemiluminescence method. We coated Streptavidin MSD plates with a biotinylated anti-VHH antibody (Genscript A01995) at 2 μg/ml in PBS for 1 hour at room temperature (RT) with 1000 rpm, washed 3X with PBT, incubated with blocking buffer for 1 hour at RT, incubated with VHH-mFc containing media/mucus (at different dilutions) for 1 hour at RT with 1000 rpm, washed 3X with PBT, incubated with ruthenylated-anti-human-Fc antibody (Clone R10Z8E9, labeled in-house) at 2 μg/ml in PBS for 1 hour at RT with 1000 rpm, washed 3X with PBT and read plates in 40 μL reading buffer using the MSD imager. The amount of VHH in basolateral and apical chambers were calculated by plotting electrochemiluminescence units (ECLU) against VHH-mFc standard curves in Prism (GraphPad).
Assessment of mucosal delivery using the epiairway model
We used the EpiAirway model, an established lung tissue model engineered from primary human tracheal bronchial cells, to test the transcytosis activity of anti-pIgR VHH-mFc molecules to the mucosal lumen. Tissue models were obtained from Mattek Corporation and maintained according to manufacturer’s instructions. 20 μg of test and control VHH-mFc molecules were added to 1 ml of EpiAirway media in the basolateral chamber and 100 μL of samples were collected from the basolateral and apical chambers at 0, 24 and 48 hours. EpiAirway TEER buffer was used to collect the mucus from the apical chambers. The amount of VHH present in basolateral media and apical mucus was quantified by electrochemiluminescence method. We coated Streptavidin MSD plates with a biotinylated anti-VHH antibody (Genscript A01995) at 2 μg/ml in PBS for 1 hour at RT with 1000 rpm, washed 3X with PBT, incubated with blocking buffer for 1 hour at RT, incubated with VHH-mFc containing media/mucus (at different dilutions) for 1 hour at RT with 1000 rpm, washed 3X with PBT, incubated with ruthenylated-anti-human-Fc antibody (Clone R10Z8E9, labeled in-house) at 2 ug/ml in PBS for 1 hour at RT with 1000 rpm, washed 3X with PBT and read plates in 40 μL reading buffer using the MSD imager. The amount of VHH in basolateral and apical chambers were calculated by plotting ECLU against VHH-mFc standard curves in Prism (GraphPad).
Confocal laser microscopy
We used indirect immunofluorescence to track the amount of pIgR and VHH-mFc retained across the EpiAirway model two-days post-treatment. We rinsed tissue samples in PBS, fixed tissues with 2 ml of 10% formalin at RT for 20 minutes, washed three times with 2 ml PBST (1% Triton-X100 in PBS) at RT, incubated with primary antibodies (500 μL apical, 500 μL basolateral) diluted in PBTG (PBST with 10% goat serum) for 2 hours at RT, washed two times with 2 ml PBTG at RT, incubated with secondary antibodies (100 μL apical, 100 μL basolateral) diluted in PBTG for 1 hour at RT and washed two times with 2 ml PBTG at RT. All wash steps were carried out for 10 minutes with gentle agitation. The primary antibody mix contained mouse anti-pIgR antibody (R&D Systems, MAB27171) and biotinylated anti-VHH antibody (Genscript A01995) both at 5 μg/ml. The secondary antibody mix contained Alexa-Flour 488-labeled anti-mouse antibody (Agilent F0479, 1:100 dilution), Alexa-Flour 647-labeled streptavidin (Invitrogen, S32357, 1:100 dilution) and Hoechst (Invitrogen H3570, 1: 1000 dilution). Fixed, permeabilized and stained tissues were imaged at 20X resolution (30–40 planes, 2 μm distance) using Opera Phenix confocal laser microscopy. Image analysis was performed using the Harmony suite, fluorescence readouts were corrected for membrane auto-fluorescence, normalized for number of cells and plotted as heat maps in Prism (GraphPad).
Cloning, expression and purification of pIgR constructs
Gene blocks-encoding desired hpIgR domain sequences were obtained from IDT and sub-cloned into mammalian expression vectors using the In-Fusion® HD Cloning Kit. pIgR domain boundaries were obtained from published literature.Citation8 HEK Expi293TM cells were transfected with pIgR-domain expression vectors using ExpiFectamineTM 293 transfection kit. Supernatants were harvested after 6–7 days by centrifugation (4,000 g, 15 min), passed through a 0.45 μm filter and purified by immobilized metal ion chromatography using HisPurTM Cobalt resin. Buffer NPI-20 was used as running buffer and Buffer NPI-300 containing 300mM Imidazole was used as elution buffer. Elution fractions were buffer-exchanged to PBS using PD10 desalting columns following manufacturer’s instructions and purified pIgR domains were stored at 4°C. Protein concentration was determined by UV-visible spectroscopy.
Characterization of VHH-mFc epitopes
The MSD platform was used for conducting epitope mapping studies. To test the binding of VHH-mFc molecules to full-length pIgR ECD and individual pIgR domains, we coated Streptavidin MSD plates with a biotinylated anti-HIS antibody (R&D systems BAM050) at 2 μg/ml in PBS for 1 hour at RT with 1000 rpm, washed 3X with PBT (PBS + 0.1% tween-20), incubated with blocking buffer for 1 hour at RT, incubated with His-tagged pIgR proteins (10 μg/ml in PBS) for 2 hours at RT with 1000 rpm, washed 3X with PBT, incubated with VHH-mFc molecules (100 μg/ml in PBS) for 2 hours at RT with 1000 rpm, washed 3X with PBT, incubated with ruthenylated-anti-human-Fc antibody (Clone R10Z8E9, labeled in-house) at 2 μg/ml in PBS for 1 hour at RT with 1000 rpm, washed 3X with PBT and read plates in 40 μL reading buffer using the MSD imager. ECLU values were plotted as a heatmap.
To show differences between VHH2 and VHH3 in binding to pIgR, we measured EC50 values for VHH-mFc molecules binding to hpIgR-ECD protein by electrochemiluminescence using two different detection antibodies, an anti-Fc antibody (Clone R10Z8E9, labeled in-house) and an anti-VHH antibody (Genscript A01995). We coated pIgR ECD (10 μg/ml in PBS) on high-bind MSD plates for 2 hours at RT with 1000 rpm, washed 3X with PBT, incubated with blocking buffer for 1 hour at RT, incubated with VHH-mFc molecules (increasing concentrations in PBS) for 1 hour at RT with 1000 rpm, washed 3X with PBT, incubated with secondary reagents (2 μg/ml in PBS) for 1 hour at RT with 1000 rpm, washed 3X with PBT and read plates in 40 μL reading buffer using the MSD imager. EC50 was calculated by fitting log VHH concentration versus log ECLU in Prism (GraphPad).
Abbreviations
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed
Competing Financial Interests Statement
All authors are employees of Janssen Research & Development, LLC.
Supplemental Material
Download Zip (4.9 MB)Acknowledgments
Authors would like to thank John Majercak and Randall Derstine for serology assessment, Mike Diem for VHH purification, Bingyuan Wu for IgA purification, Mehabaw Derebe for biophysics support and Derrick Domingo for MDCK-pIgR cells. Authors thank Jennifer Nemeth-Seay and the Postdoc program committee members for valuable discussions and comments.
Supplementary material
Supplementary information for this article can be accessed on the publisher’s website.
Additional information
Funding
Related Research Data
References
- France MM, Turner JR. The mucosal barrier at a glance. J Cell Sci. 2017;130:307–11. doi:https://doi.org/10.1242/jcs.193482.
- Brandtzaeg P, Farstad IN, Johansen FE, Morton HC, Norderhaug IN, Yamanaka T. The B-cell system of human mucosae and exocrine glands. Immunol Rev. 1999;171:45–87. doi:https://doi.org/10.1111/j.1600-065X.1999.tb01342.x.
- Leusen JH. IgA as therapeutic antibody. Mol Immunol. 2015;68:35–39. doi:https://doi.org/10.1016/j.molimm.2015.09.005.
- Hansen IS, Baeten DLP, den Dunnen J. The inflammatory function of human IgA. Cell Mol Life Sci. 2019;76:1041–55. doi:https://doi.org/10.1007/s00018-018-2976-8.
- Lewis MJ, Pleass RJ, Batten MR, Atkin JD, Woof JM. Structural requirements for the interaction of human IgA with the human polymeric Ig receptor. J Immunol. 2005;175:6694–701. doi:https://doi.org/10.4049/jimmunol.175.10.6694.
- Woof JM, Russell MW. Structure and function relationships in IgA. Mucosal Immunol. 2011;4:590–97. doi:https://doi.org/10.1038/mi.2011.39.
- Kaetzel CS. The polymeric immunoglobulin receptor: bridging innate and adaptive immune responses at mucosal surfaces. Immunol Rev. 2005;206:83–99. doi:https://doi.org/10.1111/imr.2005.206.issue-1.
- Stadtmueller BM, Huey-Tubman KE, Lopez CJ, Yang Z, Hubbell WL, Bjorkman PJ. The structure and dynamics of secretory component and its interactions with polymeric immunoglobulins. Elife. 2016;5:e10640. doi:https://doi.org/10.7554/eLife.10640.
- Bonner A, Almogren A, Furtado PB, Kerr MA, Perkins SJ. Location of secretory component on the Fc edge of dimeric IgA1 reveals insight into the role of secretory IgA1 in mucosal immunity. Mucosal Immunol. 2009;2:74–84. doi:https://doi.org/10.1038/mi.2008.68.
- Kaetzel CS. Polymeric Ig receptor: defender of the fort or Trojan horse? Curr Biol. 2001;11:R35–8. doi:https://doi.org/10.1016/S0960-9822(00)00041-5.
- Johansen FE, Kaetzel CS. Regulation of the polymeric immunoglobulin receptor and IgA transport: new advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol. 2011;4:598–602. doi:https://doi.org/10.1038/mi.2011.37.
- Lombana TN, Rajan S, Zorn JA, Mandikian D, Chen EC, Estevez A, Yip V, Bravo DD, Phung W, Farahi F, et al. Production, characterization, and in vivo half-life extension of polymeric IgA molecules in mice. MAbs. 2019;11:1122–38. doi:https://doi.org/10.1080/19420862.2019.1622940.
- Turula H, Wobus CE. The role of the polymeric immunoglobulin receptor and secretory immunoglobulins during mucosal infection and immunity. Viruses. 2018;10(5):e237. doi:https://doi.org/10.3390/v10050237.
- Bakema JE, van Egmond M, Immunoglobulin A. A next generation of therapeutic antibodies? MAbs. 2011;3:352–61. doi:https://doi.org/10.4161/mabs.3.4.16092.
- Borrok MJ, DiGiandomenico A, Beyaz N, Marchetti GM, Barnes AS, Lekstrom KJ, Phipps SS, McCarthy MP, Wu H, Dall’Acqua WF, et al. Enhancing IgG distribution to lung mucosal tissue improves protective effect of anti-Pseudomonas aeruginosa antibodies. JCI Insight. 2018;3(12):e97844. doi:https://doi.org/10.1172/jci.insight.97844.
- White KD, Capra JD. Targeting mucosal sites by polymeric immunoglobulin receptor-directed peptides. J Exp Med. 2002;196:551–55. doi:https://doi.org/10.1084/jem.20020581.
- Braathen R, Sandvik A, Berntzen G, Hammerschmidt S, Fleckenstein B, Sandlie I, Brandtzaeg P, Johansen F-E, Lauvrak V. Identification of a polymeric Ig receptor binding phage-displayed peptide that exploits epithelial transcytosis without dimeric IgA competition. J Biol Chem. 2006;281:7075–81. doi:https://doi.org/10.1074/jbc.M508509200.
- Ferkol T, Kaetzel CS, Davis PB. Gene transfer into respiratory epithelial cells by targeting the polymeric immunoglobulin receptor. J Clin Invest. 1993;92:2394–400. doi:https://doi.org/10.1172/JCI116845.
- Ferkol T, Perales JC, Eckman E, Kaetzel CS, Hanson RW, Davis PB. Gene transfer into the airway epithelium of animals by targeting the polymeric immunoglobulin receptor. J Clin Invest. 1995;95:493–502. doi:https://doi.org/10.1172/JCI117690.
- Eckman EA, Mallender WD, Szegletes T, Silski CL, Schreiber JR, Davis PB, Ferkol TW. In vitro transport of active alpha(1)-antitrypsin to the apical surface of epithelia by targeting the polymeric immunoglobulin receptor. Am J Respir Cell Mol Biol. 1999;21:246–52. doi:https://doi.org/10.1165/ajrcmb.21.2.3687.
- Ferkol T, Eckman E, Swaidani S, Silski C, Davis P. Transport of bifunctional proteins across respiratory epithelial cells via the polymeric immunoglobulin receptor. Am J Respir Crit Care Med. 2000;161:944–51. doi:https://doi.org/10.1164/ajrccm.161.3.9907018.
- Gupta S, Eastman J, Silski C, Ferkol T, Davis PB. Single chain Fv: a ligand in receptor-mediated gene delivery. Gene Ther. 2001;8:586–92. doi:https://doi.org/10.1038/sj.gt.3301451.
- Ferkol T, Cohn LA, Phillips TE, Smith A, Davis PB. Targeted delivery of antiprotease to the epithelial surface of human tracheal xenografts. Am J Respir Crit Care Med. 2003;167:1374–79. doi:https://doi.org/10.1164/rccm.200209-1119OC.
- Gupta S, Heacock M, Perez A, Davis PB. Antibodies to the polymeric immunoglobulin receptor with different binding and trafficking patterns. Am J Respir Cell Mol Biol. 2005;33:363–70. doi:https://doi.org/10.1165/rcmb.2005-0132OC.
- Emmerson CD, van der Vlist EJ, Braam MR, Vanlandschoot P, Merchiers P, de Haard HJ, Verrips CT, van Bergen En Henegouwen PMP, Dolk E. Enhancement of polymeric immunoglobulin receptor transcytosis by biparatopic VHH. PLoS One. 2011;6:e26299. doi:https://doi.org/10.1371/journal.pone.0026299.
- Ying T, Chen W, Gong R, Feng Y, Dimitrov DS. Soluble monomeric IgG1 Fc. J Biol Chem. 2012;287:19399–408. doi:https://doi.org/10.1074/jbc.M112.368647.
- O’Brien LE, Zegers MM, Mostov KE. Opinion: building epithelial architecture: insights from three-dimensional culture models. Nat Rev Mol Cell Biol. 2002;3:531–37. doi:https://doi.org/10.1038/nrm859.
- Ren D, Daines DA. Use of the EpiAirway model for characterizing long-term host-pathogen interactions. J Vis Exp. 2011;(55):e3261. doi:https://doi.org/10.3791/3261.
- Jackson GR Jr., Maione AG, Klausner M, Hayden PJ. Prevalidation of an acute inhalation toxicity test using the epiairway In Vitro human airway model. Appl In Vitro Toxicol. 2018;4:149–58. doi:https://doi.org/10.1089/aivt.2018.0004.
- Hamburger AE, West AP Jr., Bjorkman PJ. Crystal structure of a polymeric immunoglobulin binding fragment of the human polymeric immunoglobulin receptor. Structure. 2004;12:1925–35. doi:https://doi.org/10.1016/j.str.2004.09.006.
- Hamburger AE, Bjorkman PJ, Herr AB. Structural insights into antibody-mediated mucosal immunity. Curr Top Microbiol Immunol. 2006;308:173–204. doi:https://doi.org/10.1007/3-540-30657-9_8.
- Akula S, Mohammadamin S, Hellman L. Fc receptors for immunoglobulins and their appearance during vertebrate evolution. PLoS One. 2014;9:e96903. doi:https://doi.org/10.1371/journal.pone.0096903.
- Van Bockstaele F, Holz JB, Revets H. The development of nanobodies for therapeutic applications. Curr Opin Investig Drugs. 2009;10:1212–24.
- Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–97. doi:https://doi.org/10.1146/annurev-biochem-063011-092449.
- Jerdeva GV, Tesar DB, Huey-Tubman KE, Ladinsky MS, Fraser SE, Bjorkman PJ. Comparison of FcRn- and pIgR-mediated transport in MDCK cells by fluorescence confocal microscopy. Traffic. 2010;11:1205–20. doi:https://doi.org/10.1111/j.1600-0854.2010.01083.x.
- Zhang JR, Mostov KE, Lamm ME, Nanno M, Shimida S, Ohwaki M, Tuomanen E. The polymeric immunoglobulin receptor translocates pneumococci across human nasopharyngeal epithelial cells. Cell. 2000;102:827–37. doi:https://doi.org/10.1016/S0092-8674(00)00071-4.