
ABSTRACT
For antibody-drug conjugates to be efficacious and safe, they must be stable in circulation to carry the payload to the site of the targeted cell. Several components of a drug-conjugated antibody are known to influence stability: 1) the site of drug attachment on the antibody, 2) the linker used to attach the payload to the antibody, and 3) the payload itself. In order to support the design and optimization of a high volume of drug conjugates and avoid unstable conjugates prior to testing in animal models, we wanted to proactively identify these potential liabilities. Therefore, we sought to establish an in vitro screening method that best correlated with in vivo stability. While traditionally plasma has been used to assess in vitro stability, our evaluation using a variety of THIOMABTM antibody-drug conjugates revealed several disconnects between the stability assessed in vitro and the in vivo outcomes when using plasma. When drug conjugates were incubated in vitro for 24 h in mouse whole blood rather than plasma and then analyzed by affinity capture LC-MS, we found an improved correlation to in vivo stability with whole blood (R2 = 0.87, coefficient of determination) compared to unfrozen or frozen mouse plasma (R2 = 0.34, 0.01, respectively). We further showed that this whole blood assay was also able to predict in vivo stability of other preclinical species such as rat and cynomolgus monkey, as well as in human. The screening method utilized short (24 h) incubation times, as well as a custom analysis software, allowing increased throughput and in-depth biotransformation characterization. While some instabilities that were more challenging to identify remain, the method greatly enhanced the process of screening, optimizing, and lead candidate selection, resulting in the substantial reduction of animal studies.
Introduction
Antibody-drug conjugates (ADCs) typically consist of monoclonal antibodies covalently bound to cytotoxic drugs or non-cytotoxic payloads via a chemical linker.Citation1,Citation2 Combining the specific targeting capabilities of a monoclonal antibody with the cancer-killing ability of a cytotoxic drug allows ADCs to distinguish between healthy and diseased tissue and potentially widens the therapeutic window by reducing toxicities.Citation3,Citation4 Using a site-specific conjugation strategy, homogeneous ADCs with a defined drug-to-antibody ratio (DAR) can be produced by reaction at engineered cysteine residues at specific sites in antibodies without disruption of interchain disulfide bonds. ADCs with site-specific conjugation, which are also called THIOMABTM antibody-drug conjugates (TDCs), have demonstrated an improved therapeutic index in preclinical settings.Citation5,Citation6 In order to further improve the therapeutic window, a new generation of conjugates that incorporates different linkers and cytotoxic agents is being developed to improve the safety and efficacy profiles. In vitro stability studies are often used during drug discovery because they provide information that can be used to prioritize compounds for in vivo studies and alert researchers to potential liabilities of structural modifications.Citation7–Citation9 This is especially important for drug conjugates such as ADCs and TDCs, which contain linker-drugs that may be susceptible to chemical or enzymatic modifications (e.g., enzyme hydrolysis).Citation10
Drug conjugates bring the payload to the site of the tumor via the circulatory system. To be efficacious, they must retain the drug in circulation long enough to reach the targeted cell, internalize, and release the drug inside the cell to induce cell death. Therefore, understanding the stability of both the linker and drug is valuable when identifying factors for rational conjugate design. As we explore and evaluate different sites of attachment of the linker-drugs to the antibody, various linkers and a variety of drugs with alternative mechanisms of action, the ability to better predict in vivo stability in a timely manner becomes crucial. Because several components of an ADC and TDC have potential stability liabilities, early screening for these liabilities in vitro can not only reduce the number or size of in vivo studies and increase the rate at which stable conjugates are identified, but also provide important information that can help discovery teams design modified structures and interpret in vitro and in vivo efficacy experiments.Citation11–Citation14 Plasma stability has been shown to play an important role in the discovery and development of both small and large molecule drugs.Citation15,Citation16 Knowing that both the site of conjugation on the antibody and the specific linker-drug can affect stability,Citation16–Citation18 we wanted to evaluate how stability analysis, typically performed with plasma, can optimally guide drug conjugate development.
Several in vitro assays are designed to address ADC stability. Historically, we, as well as others,Citation19 have used plasma that was collected and stored frozen (this type of material is defined here as “frozen” and compared with “unfrozen” material) prior to its use evaluating the stability of specific ADCs (e.g. conjugated with the microtubule inhibitor monomethyl auristatin E (MMAE) via a cleavable linkers using maleimide chemistry). In vitro stability screening has also been done in the presence of lysosomal enzymes for multiple ADCs.Citation20 Though we have observed that in vitro plasma stability of certain MMAE ADCs translates well with in vivo outcomes, there were cases with other linker or drugs where the in vitro plasma stability did not correlate as well. In these cases, the discrepancy was either due to the amount of deconjugation of the drug from the antibody or due to a modification to the drug that was not predicted with frozen plasma but occurred in vivo. Similarly, stability screening using human liver S9 fractions showed good correlation with in vivo results for some ADCs evaluated, but the technique presented limitations related to chemical concerns for ADCs containing acid-labile linkers, as well as possible disulfide bond reduction, due to the incubation conditions. Because we wanted to support the design and optimization of novel attachment chemistries and second-generation payloads with different mechanism of actions, we sought to establish an in vitro screening method that best mimicked the stability seen in vivo for all ADCs. The goal was to ensure that not only the attachment chemistry, but also any modifications to the linker or drug, were accurately detected and would predict any unknown in vivo instabilities. Therefore, we evaluated the stability profiles of a panel of conjugates with various conjugation sites, linkers, and drug chemistries using different matrices, including frozen plasma, unfrozen plasma, and whole blood, and correlated them to in vivo stability (“unfrozen” refers to matrix collected and stored on ice before use in the assay, used within 18 h of collection, and never frozen). Here, we describe an in vitro whole blood stability assay, coupled with affinity capture liquid chromatography-mass spectroscopy (LC-MS), to characterize modifications of drug conjugates in biological matrices. With this method, we were able to reduce disconnects between in vitro and in vivo stability results, and therefore improve our ability to predict in vivo stability across multiple species. We believe that this assay provides an improved in vitro screening tool to identify stability concerns of drug conjugates with new linker-drugs, such as ADCs and TDCs, which could also be applied to other conjugated molecules. This approach has led to the improved translation of mouse stability outcomes from in vitro to in vivo, translation across other non-clinical species, and a reduction of unnecessary animal studies.
Results
Correlation of in vitro and in vivo stability in mouse
A total of 13 TDCs (Supplementary Figure 1) prepared using different chemotherapeutic agents through cleavable or non-cleavable conjugation chemistries were evaluated for stability in frozen mouse plasma. The in vitro outcomes were subsequently correlated with in vivo stability results (shown as percent drug loss over 24 h) in mice (,)). The TDCs tested were composed of five different target antibodies with payload conjugated to the same site, HC A118C,Citation21 with three different drug classes, a tubulin binder (monomethyl auristatin E, MMAE) and two DNA damaging agents (anthracycline analog, PNU; pyrrolobenzodiazepine, PBD). The drug was attached to the antibody by one of two conjugation chemistries (disulfide or maleimide) using either non-cleavable or cleavable linkers where the linker is stable under a wide variety of conditions but can be efficiently and selectively cleaved by either reduction, proteolysis, or acid. TDCs were incubated in plasma for up to 96 h at 37°C, followed by affinity capture LC-MS analysis. An MS software analysis tool was customized to assess the in vitro stability of the TDCs, and the percent loss or modification of drug, relative to the 0 h time-point, were compared with in vivo stability. A lack of correlation was observed between the in vivo stability and the stability in frozen plasma at 24 h (R2= 0.01) ()) and 96 h (R2= 0.0002) ()). Using unfrozen plasma, the correlation, although slightly improved, was still poor at the 24 hr (R2= 0.34) ()) and 96 hr (R2= 0.03) time-points ()). Extending the incubation in plasma from 24 h (frozen R2 = 0.01, unfrozen R2 = 0.34) to 96 h (frozen R2 = 0.0002, unfrozen R2 = 0.03) made the correlation to the in vivo results at matching time-points worse.
Figure 1. Lack of in vitro plasma stability translation to in vivo results. Correlation of mouse in vivo stability with in vitro stability for 13 TDC conjugates in frozen mouse plasma (a and b) and unfrozen mouse plasma (c and d) at 24 hr or 96 hr where 0 = no loss (stable) and 100 = complete loss (unstable); “loss” = linker-drug deconjugation and/or inactivating payload metabolism. Axes represent percent loss of drug relative to 0 h. (e, f) Specific examples of mouse plasma stability of MS spectra DAR profiles (top, middle) showing disconnect with in vivo stability (bottom) for an anti-HER2 TDC conjugated with an anthracycline analog (PNU) (e) and an anti-HER2 TDC conjugated to MMAE (f). MS peak labels indicate an antibody (Ab, glycated (Glc)) with one linker drug (+ LD) or two (+ 2*LD) can have drug lost and replaced by a cysteine (+ Cys) or a glutathione (+ GSH) or modified (-E)
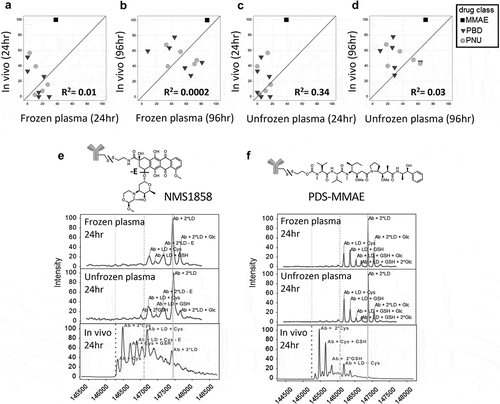
We observed several in vitro plasma stability disconnects for TDCs compared to in vivo stability when using −80°C stored plasma or unfrozen plasma. ) shows the deconvoluted mass spectra stability profiles of two TDCs incubated in mouse plasma (frozen and unfrozen) for 24 h compared to in vivo at the same time-point. Results shown in ) indicate that an anti-human epidermal growth factor receptor 2 (HER2) TDC conjugated to an anthracycline analog drug via a disulfide linker (anti-HER2 7C2 HC A118C NMS-1858) was more stable in mouse plasma at 24 h compared to in vivo. The loss of drug from the antibody and loss of part of the drug (-E, ether cleavage) occurred to a much greater extent in vivo compared to in vitro when using plasma. Similarly, as shown in ), an anti-HER2 TDC conjugated to MMAE with a disulfide linker (anti-HER2 4D5 HC A118C PDS-MMAE) demonstrated greater stability in both frozen and unfrozen plasma at 24 h compared to in vivo, where greater drug loss from the antibody was observed.
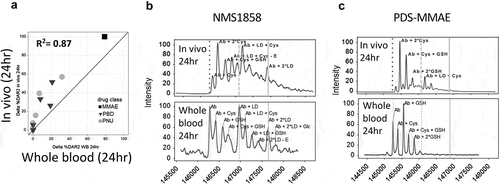
To overcome these disconnects, we sought to develop a whole blood stability assay with improved correlation to in vivo stability by obtaining whole blood from mouse (as well as rat, cynomolgus monkey [cyno] and human), and incubating the same 13 TDCs for up to 24 h at 37°C, followed by affinity capture LC-MS analysis (Supplementary Figure 2). This assay greatly improved the correlation between in vitro and in vivo stability of the TDC at 24 h for mice (R2= 0.87) ()) compared to the frozen and unfrozen plasma (R2= 0.34, 0.01, respectively; )). The extent of drug deconjugation and drug modification seen in whole blood was more similar to the TDC stability seen in vivo for both the anti-HER2 7C2 HC A118C NMS-1858 and anti-HER2 4D5 HC A118C PDS-MMAE ()) compared to plasma ()).
Figure 2. Improved correlation of in vitro stability with in vivo stability using whole blood. Correlation of TDC in vivo stability with unfrozen whole blood compared to in vivo at 24 h (a) using the same 13 conjugates evaluated in plasma (). (b, c) Specific examples of MS spectra of in vivo DAR stability profiles (B, C bottom) compared to stability profile in whole blood (top) for an anti-HER2 TDC conjugated with an anthracycline analog (PNU) (b) and an anti-HER2 TDC conjugated to MMAE (c). Axes in (A) represent the loss of drug percentage relative to 0 hr. MS peak labels indicate an antibody (Ab, glycated (Glc)) with one linker drug (+ LD) or two (+ 2*LD) can have drug lost and replaced by a cysteine (+ Cys) or a glutathione (+ GSH) or modified (-E). A 96 h time-point in whole blood was not evaluated because, after 48 h at 37°C, the samples could not be processed due to clotting of the whole blood
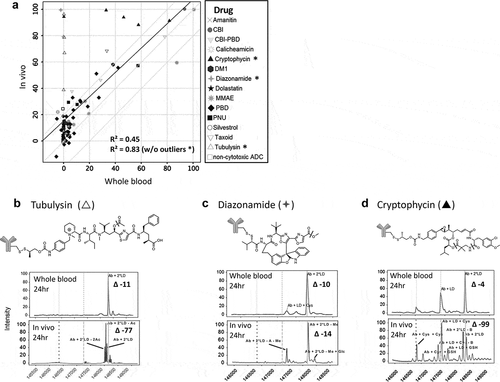
Additional TDCs were evaluated for stability in whole blood for a total of 98 different conjugates (14 different cytotoxic drugs plus 2 non-cytotoxic drugs conjugated at different sites to various antibodies with different linkers) and correlated with in vivo stability at 24 h ()). For the sites evaluated, the antibody target did not significantly affect the stability profile of the linker or drug. The overall R2 value for the in vitro whole blood and in vivo stability correlation at 24 h with this larger sample set was 0.45. The stability outliers identified consisted of three drugs (tubulysin, diazonamide, and cryptophycin), where a drug modification occurred to a lesser extent in the whole blood than in vivo ()). The R2 value of the correlation in the absence of these 3 drugs improved to 0.83 with an n = 88. To address these disconnects, the impact of pH or pH change during incubation in the biological matrices was evaluated (Supplementary Figure 3), and showed that there was only a slight difference or change in pH in plasma and whole blood; however, plasma samples became slightly more basic while whole blood samples became slightly more acidic. The pH difference after 24 h between plasma and whole blood was >0.5. Based on preliminary data (not shown), this difference in pH did not impact the stability profile of the outliers.
Figure 3. Identification of stability outliers between mouse in vitro whole blood and in vivo outcomes. Additional TDC conjugates were evaluated for stability and their in vitro and in vivo correlation compared (a). TDC conjugates outside of the ± 20% (dashed line) of linear regression line (solid line) were identified as outliers and spectra are shown (B, C, D). Axis in (A) represents a change in DAR2 at 24 h in matrix relative to buffer at 0 hr. MS peak labels indicate an antibody (Ab, glycated (Glc)) with one linker drug (+ LD) or two (+ 2*LD) can have drug lost and replaced by a cysteine (+ Cys) or a glutathione (+ GSH) or modified (acetyl loss – Ac, methyl loss – Me, amide and ester hydrolysis – B)
Stability translation across other species and species-dependent differences
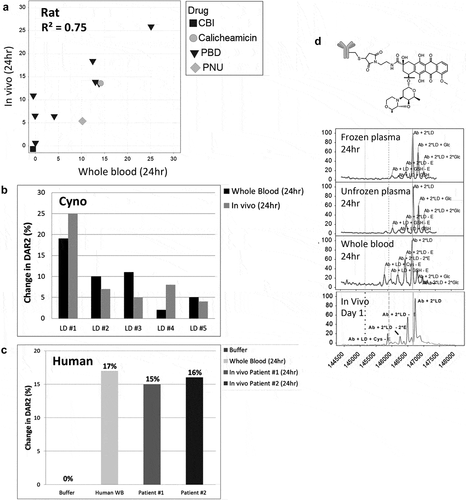
In addition to mouse, which is primarily used as an efficacy model for ADCs, we also investigated the translation of in vitro to in vivo stability using the whole blood assay for rat, cyno, and human ()), as these species are utilized in toxicity and pharmacokinetics (PK) studies. We observed relatively similar stabilities in vitro and in vivo for all three species, where the change in DAR2 was within ±10%. For rat ()), where we had a larger data set (n = 11), the R2 was 0.75. While we had fewer data points for cyno ()), the in vitro and in vivo stabilities were within ±6% change in DAR2 of each other for the 5 conjugates evaluated (PBD TDCs with different linkers). Although the number of samples was limited, using in vivo samples ()) from humans dosed with a TDC bearing the microtubule inhibitor MMAE, we observed similar stability profiles for two different patients compared to our in vitro data at 24 h. Although whole blood stability correlated with in vivo stability, we were able to observe a stability disconnect between plasma and whole blood for cyno with a TDC conjugated to an anthracycline analog (PNU) drug (anti-HER2 4D5 HC A118C NMS-03123048) ()). Similar to the analysis for mice, this TDC was more stable in both frozen and unfrozen cyno plasma after 24 h compared to 24 h in whole blood where both drug deconjugation and drug modification were observed. This result better matched what was seen in vivo at 24 h. Because this drug modification (ether cleavage) inactivates the payload and thereby affects the drug load (DAR), the correlation of DAR loss also improved when performing stability evaluation using whole blood compared to plasma.
Figure 4. In vitro to in vivo stability translation in other species. Correlation of in vitro TDC stability with in vivo stability in (a) rat (top), (b) cyno, and (c) human (bottom). Evaluated 11 conjugates for rat, 5 pyrrolobenzodiazepine conjugates with different linkers for cyno and 1 conjugate for human. (b) Specific example of a cyno plasma stability disconnect with cyno whole blood and in vivo for a TDC with an anthracycline analog. MS peak labels indicate an antibody (Ab, glycated (Glc)) with one linker drug (+ LD) or two (+ 2*LD) can have drug lost and replaced by a cysteine (+ Cys) or a glutathione (+ GSH) or modified (ether cleavage – E)
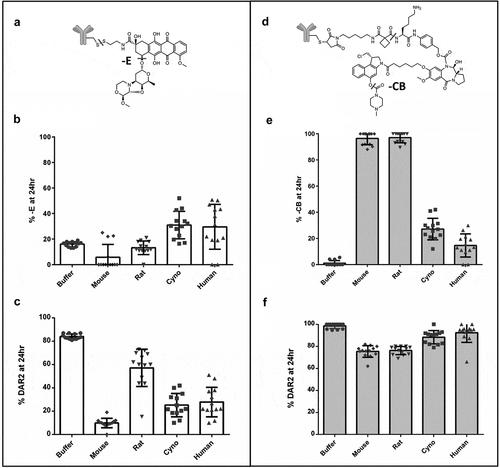
In the process of screening TDCs across multiple species (mouse, rat, cyno, human), we observed several species-dependent stabilities. Two examples of TDCs with these species-dependent stability differences are shown in ,). These conjugates were also used as assay controls to monitor different shipments of whole blood as they each had two possible modifications. One of these example conjugates, identified to be more unstable in primate matrix compared to rodents, was the anthracycline analog drug shown in ). The instability due to ether cleavage (labeled – E), which leads to loss of the sugar group, is seen to a higher extent in the cyno and human matrix compared to the rodent species after 24 h ()). The second example is a TDC conjugated with a CBI-PBD heterodimer containing a carbamate prodrug as shown in ). The instability of the carbamate prodrug is much more evident in rodents after 24-h relative to primates ()). Even earlier time-points (data not shown) showed differential stability across the species due to different rates of carbamate loss. ,) shows the impact of deconjugation and drug modification on the overall activity/DAR of the two TDCs.
Figure 5. Reproducibility and species difference of whole blood stability for two control TDCs (a, b) using whole blood shipped overnight (n = 13). Differences in drug modification (b ether cleavage (-E), e carbamate hydrolysis (-CB)) and DAR (c disulfide cleavage, f maleimide cleavage) between buffer and four species for two different conjugates (b, c and e, f) run as controls for each shipment of whole blood
Reproducibility in whole blood
The two examples of species-dependent differences were used to evaluate the reproducibility of the assay with different lots of whole blood from independent overnight shipments (); ). These quality control standards had unique stabilities and were used to monitor the performance of the assay from run to run. Each control TDC had a different stability profile resulting from deconjugation of the linker drug at the site of antibody attachment (either disulfide or maleimide exchange) and a specific drug modification (either ether cleavage or carbamate loss). The control TDCs showed consistent species- and molecule-dependent stability differences between runs from different lots of whole blood (n = 13), suggesting good reproducibility of the assay process (within ±20% DAR2 loss or drug modification). The average percent modification and DAR changes for both controls, along with their standard deviations, are listed in . Greater variability in drug loss (change in DAR) was seen for control #1 where the deconjugation and drug modification (ether cleavage) both resulted in drug inactivation. Control #2 was less variable for drug loss, as the drug modification (carbamate loss) did not result in drug inactivation. The age ranges of the different species, which were 6–8 weeks for rodents, 2–14 years for cynomolgus monkeys and 30–65 years for humans, are also included in . The mouse strain used in this screening assay was CB17 SCID Beige, the rats were Sprague Dawley, the monkeys were cynomolgus of primarily Chinese, with some of Mauritius or Indonesian origins, and the humans were of either African or Hispanic ethnicities. Rodent samples were from a pool of animals (mice n = 9 and rats n = 2), while cyno and human samples were from individuals.
Table 1. Differences in DAR and drug modification between buffer and matrix from four species for two different conjugates run as controls with each shipment of whole blood
We also evaluated potential variability from one animal to another in the same study by comparing the stability of three different TDCs (anti-HER2 NMS-36249 TDC, anti-HER2 4D5 HC A118C SG-3244 TDC, and anti-HER2 4D5 HC A118C MC-vc-PAB-MMAE TDC) in whole blood from three individual rats and three individual cynomolgus monkeys (Supplementary Figure 4–6). Data showed that the stability profile of the three TDCs, as seen from the deconvoluted spectra, were relatively similar for all three individual rats or cynos. The % DAR2 at 24 h was within ± 10% for all the three TDCs in both rat and cyno whole blood (Supplementary Figure 7). Greater reproducibility was seen with these conjugates, which can only have a single modification compared to conjugates that can have two separate modifications ()), as is the case with the assay controls.
Discussion
In our effort to support the design and optimization of novel attachment chemistries and second-generation payloads with different mechanism of actions, we wanted an in vitro stability screening strategy that could identify and exclude unstable molecules before in vivo studies were done. When using our in vitro plasma stability assay, we discovered several stability disconnects with in vivo outcomes related to either the deconjugation of the linker drug from the antibody or the modification of the payload, or both. Given the liabilities of the in vitro plasma stability assay, we sought to evaluate the utility of in vitro whole blood assessments as a way to improve our ability to predict in vivo stability of TDCs. When using whole blood for the in vitro screen, we were able to show an improved correlation to in vivo outcomes, which allowed us to confidently eliminate unstable TDCs before in vivo studies were initiated.
While the in vitro whole blood assay was very successful at screening out unstable molecules, we observed a few stability differences where the in vitro stability assay was unable to predict in vivo liabilities. There are various factors that could contribute to the stability disconnects between plasma, whole blood, and in vivo samples. Different levels of cysteine or glutathione between whole blood and plasma could play a role in disconnects observed for disulfide-linked drugs. For example, storage conditions of plasma have little influence on homocysteine values,Citation22 while in whole blood, an increase of homocysteine is observed after collection because of ongoing metabolism and time-dependent release from erythrocytes.Citation23–Citation25 It has also been reported that glutathione, cysteinylglycine, and γ-glutamylcysteine show an increase over time if whole blood is stored due to red blood cell lysis, whereas cysteine decreases.Citation24,Citation26 Preliminary data using glutathione spiked into buffer reproduced the instability of a disulfide-linker TDC (SG3244 TDC) observed in vivo (data not shown). Additional components affected by storage, according to Oddoze et al.,Citation27 are potassium, inorganic phosphorus, magnesium, LD, glucose, lactate, mean corpuscular volume, mean corpuscular hemoglobin, activated partial thromboplastin time, insulin, C-peptide, PTH, osteocalcin, C-telopeptide, and ACTH. Differences between stability in plasma/serum and stability in whole blood for some well-defined classes of (small) molecules such as N-oxides and hydroxamic acids after short incubations have also been reported.Citation28–Citation32
Another difference between the matrices may be pH. Di et al.Citation14 reported that plasma pH increases to pH 8–9 during long-term storage at −80°C, while the in vivo pH is approximately 7.4. Our measurements indicated that the plasma pH was just above 7.5 after 24 h incubation, while whole blood was just slightly below pH 7. This subtle difference in pH could result in different rates of hydrolysis and may explain the disconnects between whole blood and in vivo ()). We therefore performed additional stability analysis with pH-adjusted whole blood to mimic in vivo conditions, but our preliminary data showed no change to the stability profile, and we did not observe an increase in the drug modifications (data not shown).
Although plasma can be conveniently frozen and stored at −80°C, while whole blood cannot, the freezing of plasma can also reduce any enzymatic activity responsible for modifying the linker-drug components. Examples of disconnects potentially related to enzymatic differences have been reported by Dorywalska et al.Citation33,Citation34 They noticed that in mice the VC-PABC linker appears to be cleaved more efficiently in vivo relative to the in vitro assay. They rationalized that this could be a result of a continuous source of the putative enzyme(s) in vivo, or due to a contribution from intracellular cleavage and release of the unconjugated antibody through endocytic recycling. They showed that compounds with certain functional groups, including esters, amides, lactones, lactams, carbamides, sulfonamides, and peptide mimetics, are more susceptible to hydrolysis by plasma enzymes. Di et al. also showed that drugs containing ester or amide functional groups are susceptible to hydrolysis by plasma enzymes.Citation3,Citation13,Citation14 This is consistent with our findings that the stability of payloads susceptible to deacetylation or ester hydrolysis were more difficult to predict in our whole blood in vitro assay.
Greater stability variability may also reflect differences in enzyme or molecule levels across species. For example, it is known that high levels of carboxylesterases are in mouse circulation relative to other species,Citation3,Citation10,Citation33,Citation34 and that carboxylesterases are only abundant in rabbit and rodent plasma, but are not detectable in primate and dog plasma.Citation35 As a result, the species difference of the carboxylesterases level in the plasma can lead to a marked difference in stability in animal species, as seen in . There have also been some reported differences in rat cysteine and methionine levels. Stabler et al. showed that cysteine was significantly (P < .05) higher in human serum than in rat serum and methionine was significantly (P < .05) higher in rat serum than in human serum.Citation23 Additional stability variability may be related to the challenges in implementing the whole blood stability assay as a primary screen, which included logistics of shipping whole blood and reproducibility of the assay from batch to batch of the blood to ensure good correlation of in vitro stability to in vivo efficacy studies.
Although our method successfully identified a number of stability liabilities, due to the resolution and sensitivity of the mass spectrometer used we may not have measured all possible modifications. Since the drug losses are identified according to the corresponding mass shifts from the starting material using a quadrupole time-of flight mass spectrometer with a resolution limitation of 10 Da, resolving peaks that were close together was challenging. It is also possible that low levels of partial proteolysis could go unnoticed due to the heterogeneous peaks generated. Both of these limitations can be overcome by analyzing the stability samples on a mass spectrometer with higher resolution and greater sensitivity.
In conclusion, to better identify liabilities that can negatively impact the stability of TDCs prior to testing in animal models, we developed an in vitro stability assay with improved correlation to in vivo stability by substituting plasma with whole blood. Although whole blood has been used for stability analysis of small molecules for short incubations, we were able to use it for improved stability analysis of TDC molecules for up to 24 h. For mice, an improved correlation was observed between the in vitro and in vivo stability of the TDC at 24 h in whole blood compared to frozen and unfrozen plasma. Our whole blood stability screening approach not only showed improved translation of stability outcomes from in vitro to in vivo for TDCs with an extensive variety of payloads and linkers for mouse stability, but the simultaneously profiling of stability across multiple species, including rat, cyno, and human, allowed additional prioritization and alerted us to potential liabilities. Similarly to predicting stability in mouse, the whole blood assay was able to predict in vivo stability in rat, cyno, and human. Although the stability of payloads susceptible to deacetylation or ester hydrolysis were more difficult to predict, our whole blood stability assay showed improved translation to in vivo compared to plasma, and enhanced our ability to screen, optimize, and prioritize a large number of TDCs. Moreover, in order to analyze the stability of a large number of conjugates, a custom MS data analysis tool was developed to visualize deconvoluted spectra, automatically label peaks, and aggregate peak data for DAR conversion. This established and streamlined the whole blood stability approach, enabling us to greatly minimize the number of in vivo studies and confirm that stabilities translated across all preclinical species. The screening strategy of using whole blood across multiple species to improve the translation of in vivo stability can be applied to any molecule that can be affinity captured and analyzed by MS, expanding the utility of this platform.
Materials & methods
Materials
Plasma and whole blood (CB17 SCID mouse, Sprague-Dawley rat, cynomolgus monkey, and human) were purchased from BioIVT (Westbury, NY). Streptavidin (SA)-coated Dynabeads M-280 were purchased from Thermo Fisher Scientific (catalog #60210), Peptide N-glycosidase F (PNGase F) was from New England Biolabs (catalog #P0704B) and HBS-EP buffer containing 0.01 M HEPES, pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.005% Polysorbate 20 (GE Healthcare Life Sciences, catalog #BR-1001-88). All specific TDC capture reagents (i.e., extracellular domain [ECD] or anti-idiotypic antibody) were produced at Genentech (South San Francisco, CA). Anti-HER2 monoclonal antibody (Herceptin®, trastuzumab) and TDC compounds were generated in-house from Genentech (South San Francisco, CA). TDCs were generated by conjugation of the antibody as previously described.Citation6
Preparation of TDCs
In general, conjugates were prepared by reacting to the two engineered Cys residues on THIOMABTM antibody-drug conjugates with activated disulfide analogs of the PBD linker-drugs. Antibodies were produced in Chinese hamster ovary cells and purified using standard methods. In these antibodies, the engineered Cys residues are present as mixed disulfides with cysteine or glutathione, which must be removed (“deblocked”) to enable conjugation to the engineered Cys sulfhydryl groups. To accomplish this, the antibodies were partially reduced with dithiothreitol, purified, reoxidized with dehydroascorbic acid and purified again into a succinate buffer (10 mM succinate, pH 5.0, 150 mM NaCl, 2 mM ethylenediaminetetraacetic acid (EDTA)) to give the deblocked THIOMABTM antibody-drug conjugates. The pH of the deblocked antibody was adjusted with 1 M Tris, pH 8.5 (75 mM final Tris concentration) and 3 equivalents of nitropyridyl disulfide PBD linker-drugs (10 mM stock in dimethylformamide) added. The conjugation reactions were allowed to proceed to completion at room temperature as indicated by LC-MS analysis of the reaction mixture (3–4 h). Reaction mixtures were diluted 5-fold into 20 mM histidine-acetate, pH 5.5 buffer and loaded onto S maxi strong-cation exchange columns (Pierce, catalog #90009), washed several times with histidine-acetate and eluted in 20 mM histidine-acetate, pH 5.5, 300 mM NaCl. Conjugates were formulated into 20 mM histidine-acetate, pH 5.5, 240 mM sucrose by dialysis. Conjugates were analyzed for DAR by reverse-phase LC-MS (PLRP-S column, Agilent TOF instrument), for aggregation by analytical size exclusion chromatography (Shodex column, Agilent HPLC instrument) and for endotoxin by using a limulus amebocyte lysate assay by Charles River Laboratories. All conjugates had DAR values of 1.8–2.0, were >98.5% monomeric and had endotoxin levels <0.1 EU/mg.
Biotinylation of capture reagents
Capture reagents (i.e., target antigens) were biotinylated with NHS-PEG4-Biotin using a commercial kit according to manufacturer instructions (Thermo Fisher Scientific, catalog #21329). Briefly, capture reagents in phosphate-buffered saline (PBS, pH 7.4) were incubated with a 10-fold molar excess of 20 mM of NHS-PEG4-Biotin for 30 min at room temperature. A NAP-5 column (GE Healthcare Life Sciences, catalog #17-0853-02) was equilibrated with 10 ml PBS (pH 7.4)), loaded with the biotinylated capture reagents, and eluted by 1 ml of PBS buffer to remove the nonreacted NHS-PEG4, as per the manufacturer’s protocol. Biotinylated capture reagents concentrations were determined spectrophotometrically by measuring the absorbance at 280 nm using a NanoPhotometer (Implen, Westlake Village, CA)
Plasma and whole blood incubation
Matrix collected in lithium heparin-containing tubes (rather than EDTA so as not to inhibit any enzymes in the biological matrix) was shipped by the vendor (BioIVT, Westbury NY). Unfrozen plasma and whole blood were collected in the afternoon and shipped cold (2-8°C) overnight so as to arrive within 18 h of collection, while frozen plasma was collected and shipped frozen with normal delivery conditions. For unfrozen plasma and whole blood, samples were created immediately upon arrival. TDC source material was made to 1mg/mL in Buffer (1X PBS [pH 7.4], 0.5% bovine serum albumin, 15 parts per million ProclinTM) and then further diluted to a final concentration of 100 ug/mL. Once mixed, 150 μL of the whole blood/Buffer stability samples was aliquoted into two separate sets of tubes for the two different time-points. The 0 h time-points were then placed in a −80°C. Whole blood or plasma stability samples were created (n = 1, 4 matrices + buffer, 100 ug/mL TDC) with 3 aliquots of 150 uL for 0, 24, and 96 h time-points for plasma and two aliquots of 150uL for 0 and 24 h time-points for whole blood. The 0 h samples were placed in a −80°C freezer and 24 and 96 h samples were shaken (~700 rpm) in 37°C incubator. Aliquots were collected at 24 and 96 h and stored in −80°C freezer until affinity capture LC-MS was performed. The matrices used to generate the samples were mouse (CB17 SCID), rat (Sprague-Dawley), monkey (cynomolgus) and human. For these incubations, we did not control the pH.
In vitro stability sample analysis
The 0, 24 or 96 h plasma and 0 or 24 h whole blood stability samples were analyzed by affinity-capture LC-MS with modifications to the method described previously.Citation15 Briefly, SA-coated magnetic beads (Thermo Fisher Scientific, catalog #60210) were washed 2x with HBS-EP buffer (GE Healthcare Life Sciences, catalog #BR-1001-88), then mixed with either biotinylated extracellular domain of target (e.g., human HER2) or anti-idiotypic antibody for specific capture or biotinylated human IgG for generic capture using a KingFisher Flex (Thermo Fisher Scientific) and incubated for 2 h at room temperature with gentle agitation. The SA-bead/Biotin-capture probe complex was then washed 2x with HBS-EP buffer, mixed with stability samples pre-diluted 1:16 with HBS-EP buffer and incubated for 2 h at room temperature with gentle agitation. After the 2 h, the SA-bead/Biotin-capture probe/sample complex was washed 2x with HBS-EP buffer, and then deglycosylated via overnight incubation with PNGase F (New England Biolabs, catalog #P0704B). The SA-bead/Biotin-capture probe/sample complex was then washed 2x with HBS-EP buffer, followed by 2x washes of water (Optima™ LC/MS Grade, Fisher Chemical, catalog #W6-1) and finally 1x wash with 10% acetonitrile. The beads were placed in 30% acetonitrile/0.1% formic acid for elution for 30 min at room temperature with gentle agitation before the beads were collected. The eluted samples were then loaded on to the LC-MS (Synapt-G2S, Waters, Milford, MA) for analysis.
For in vitro stability samples, 10 μL of TDC samples was injected and loaded onto a PepSwift RP monolithic column (500 µm × 5 cm) (Thermo Fisher Scientific, catalog #164585) maintained at 65°C. The TDC was separated on the column using a Waters Acquity UPLC system at a flow rate of 20 µL/min with the following gradient: 20% B (95% acetonitrile + 0.1% formic acid) at 0–2 min; 35% B at 2.5 min; 65% B at 5 min; 95% B at 5.5 min; 5% B at 6 min. The column was directly coupled for online detection with a Waters Synapt G2-S Q-ToF mass spectrometer operated in positive electrospray ionization mode with an acquisition mass range from m/z 500 to 5000 Da.
For stability data analysis, the deconvolution of the raw spectrum within a selected TDC elution time window was implemented with Waters BiopharmaLynx 1.3.3 software. The drug loss or modifications were identified according to the corresponding mass shifts from the starting TDC material. Peak labeling and % DAR calculation were performed with a custom Vortex script (Dotmatics, Bishops Stortford, United Kingdom). Drug loss, cleavage or adducts formation were identified according to the corresponding mass shifts from the starting TDC material. The relative abundance of each TDC species in the analytical sample was represented by its MS signal intensity. The relative ratios of TDC with different DARs were calculated by dividing the intensity of the specific TDC species with the intensity from the total TDC species. % DAR was calculated as previously reported.Citation15
In vivo studies
All animal studies were carried out in compliance with the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee at Genentech, Inc.
For in vivo stability analysis from mouse efficacy and PK studies, whole blood was collected from selected animals of different strains (including nu/nu, C.B-17 SCID, SCID-beige from Charles River Laboratories). Animals were given a single intravenous dose of ADCs and blood samples (n = 2-3/time-point) were collected at the indicated time post-dose. To minimize the stress that might influence animal health, blood volume is limited to 0.15 mL/animal and sample-collection times are rotated between animals so each animal is bled no more than twice within a week. Blood samples were collected into tubes containing lithium heparin and allowed to sit on wet ice until centrifugation (within 15 min of collection). Samples were centrifuged at 10,000 rpm for 5 min at 4°C. Plasma was then collected, placed on dry ice, and stored at −70°C until analysis.
Blood samples collected from rat and cynomolgus monkey PK studies used for in vivo stability analysis were collected from the tail vein (rat) or the saphenous vein (cynomolgus monkey) at the indicated time post-dose. Blood samples were collected into tubes containing lithium heparin and allowed to sit on wet ice until centrifugation (within 15 min of collection). Samples were centrifuged at 10,000 rpm for 5 min at 4°C. Plasma was then collected, placed on dry ice, and stored in a freezer set to maintain a temperature range of −68°C to −72°C until analysis.
In vivo stability sample analysis
Representative in vivo samples were analyzed for the stability of TDCs by either an affinity capture LC-MS assay,Citation15 or its improved version, affinity capture LC-MS F(ab’)2 assay.Citation18 Briefly, in the affinity capture LC-MS assay, biotinylated capture reagents, e.g., the ECD of a receptor, were immobilized onto SA paramagnetic beads at room temperature. TDC-containing plasma samples were incubated with the above ECD-immobilized beads to allow immunocapture of TDCs. The intact TDC complex was then deglycosylated with PNGase F at 37°C overnight and eluted from beads prior to LC-MS analysis. Alternatively, to improve the sensitivity and resolution of the analysis, captured TDCs could undergo digestion by IdeS (Genovis, Inc., catalog #A0-FR1-020) at 37°C for 1 h instead to generate the smaller F(ab’)2 fragments, which were subsequently eluted and injected onto LC-MS. The LC-MS analysis was typically performed on an AB Sciex TripleTOF 5600 mass spectrometer coupled with a Waters nanoACQUITY UPLC system. Details on the experimental setup can be found in previous publications.Citation15,Citation18
Given the slightly different assay conditions used for evaluating in vivo and in vitro stability (i.e., deglycosylation enzyme and elution buffers), controls were run using both methods and no difference in stability results were observed (data not shown).
Supplemental Material
Download MS Word (1.7 MB)Acknowledgments
The authors thank Daniel Tran and Ryan Cook for project support as well as Susan Spencer, Rachana Ohri, Genee Lee, Becca Rowntree, and Thu Anh Nguyen for reagent organization and distribution. We also thank the Genentech Research Conjugation Lab and WuXi Apptec for generating the TDCs and Douglas Leipold, Shabkhaiz Masih, Brandon Latifi, Isabel Figueroa, Norman Zhou, Kirsten Achilles Poon, Brian Vuillemenot, Donna Lee, Nicola Stagg, Fiona Zhong, Jeffrey Lau, Geoffrey del Rosario, MaryAnn Go, Sharon Yee for performing animal studies and generating in vivo samples. All authors except for Aimee Fourie-O’Donohue and Robert Tchelepi, are full-time employees of Genentech, a member of the Roche Group, and have been granted options and own shares of Roche Holding AG.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Related Research Data
References
- Chari RVJ, Miller ML, Widdison WC. Antibody–drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl. 2014 Apr 7;53(15):3796–11. doi:https://doi.org/10.1002/anie.201307628.
- Peters C, Brown S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep. 2015 Jun 12;35(4). doi:https://doi.org/10.1042/BSR20150089.
- Wang H, Rangan VS, Sung MC, Passmore D, Kempe T, Wang X, Thevanayagam L, Pan C, Rao C, Srinivasan M, et al. Pharmacokinetic characterization of BMS-936561, an anti-CD70 antibody-drug conjugate, in preclinical animal species and prediction of its pharmacokinetics in humans. Biopharm Drug Dispos. 2016 Mar;37(2):93–106. doi:https://doi.org/10.1002/bdd.1953.
- Polakis P. Antibody drug conjugates for cancer therapy. Pharmacol Rev. 2016 Jan;68(1):3–19. doi:https://doi.org/10.1124/pr.114.009373.
- Strop P, Delaria K, Foletti D, Witt JM, Hasa-Moreno A, Poulsen K, Casas MG, Dorywalska M, Farias S, Pios A, et al. Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat Biotechnol. 2015 Jul;33(7):694–96. doi:https://doi.org/10.1038/nbt.3274.
- Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008 Aug;26(8):925–32. doi:https://doi.org/10.1038/nbt.1480. Epub 2008 Jul 20.
- Borthwick AD, Exall AM, Haley TM, Jackson DL, Mason AM, Weingarten GG. Pyrrolidine-5,5-trans-lactams as novel mechanism-based inhibitors of human cytomegalovirus protease. Part 3: potency and plasma stability. Bioorg Med Chem Lett. 2002 Jul 8;12(13):1719–22. doi:https://doi.org/10.1016/S0960-894X(02)00294-9.
- Borthwick AD, Davies DE, Ertl PF, Exall AM, Haley TM, Hart GJ, Jackson DL, Parry NR, Patikis A, Trivedi N, et al. Design and synthesis of pyrrolidine-5,5-trans-lactams (5-oxohexahydropyrrolo[3,2-b]pyrroles) as novel mechanism-based inhibitors of human cytomegalovirus protease. 4. Antiviral activity and plasma stability. J Med Chem. 2003 Oct 9;46(21):4428–49. doi:https://doi.org/10.1021/jm030810w.
- Breitenlechner CB, Wegge T, Berillon L, Graul K, Marzenell K, Friebe WG, Thomas U, Schumacher R, Huber R, Engh RA, et al. Structure-based optimization of novel azepane derivatives as PKB inhibitors. J Med Chem. 2004 Mar 11;47(6):1375–90. doi:https://doi.org/10.1021/jm0310479.
- Dorywalska M, Dushin R, Moine L, Farias SE, Zhou D, Navaratnam T, Lui V, Hasa-Moreno A, Casas MG, Tran TT, et al. Molecular basis of valine-citrulline-PABC linker instability in site-specific ADCs and its mitigation by linker design. Mol Cancer Ther. 2016 May;15(5):958–70. doi:https://doi.org/10.1158/1535-7163.MCT-15-1004.
- Kerns EH. High throughput physicochemical profiling for drug discovery. J Pharm Sci. 2001 Nov;90(11):1838–58. doi:https://doi.org/10.1002/jps.1134.
- Kerns EH, Di L. Pharmaceutical profiling in drug discovery. Drug Discov Today. 2003 Apr 1;8(7):316–23. doi:https://doi.org/10.1016/S1359-6446(03)02649-7.
- Di L, Kerns EH. Profiling drug-like properties in discovery research. Curr Opin Chem Biol. 2003 Jun;7(3):402–08. doi:https://doi.org/10.1016/S1367-5931(03)00055-3.
- Di L, Kerns EH, Chen H, Petusky SL. Development and application of an automated solution stability assay for drug discovery. J Biomol Screen. 2006 Feb;11(1):40–47. doi:https://doi.org/10.1177/1087057105281363. Epub 2005 Oct 18.
- Xu K, Liu L, Saad OM, Baudys J, Williams L, Leipold D, Shen B, Raab H, Junutula JR, Kim A, et al. Characterization of intact antibody-drug conjugates from plasma/serum in vivo by affinity capture capillary liquid chromatography-mass spectrometry. Anal Biochem. 2011 May 1;412(1):56–66. doi:https://doi.org/10.1016/j.ab.2011.01.004.
- Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol. 2012 Jan 22;30(2):184–89. doi:https://doi.org/10.1038/nbt.2108.
- Ohri R, Bhakta S, Fourie-O’Donohue A, Dela Cruz-Chuh J, Tsai SP, Cook R, Wei B, Ng C, Wong AW, Bos AB, et al. High-throughput cysteine scanning to identify stable antibody conjugation sites for maleimide- and disulfide-based linkers. Bioconjug Chem. 2018 Feb 21;29(2):473–85. doi:https://doi.org/10.1021/acs.bioconjchem.7b00791.
- Su D, Ng C, Khosraviani M, Yu S, Cosino E, Kaur S, Xu K. Custom-designed affinity capture LC-MS F(ab′)2 assay for biotransformation assessment of site-specific antibody drug conjugates. Anal Chem. 2016 Dec 6;88(23):11340–46. doi:https://doi.org/10.1021/acs.analchem.6b03410.
- Durbin KR, Nottoli MS, Catron ND, Richwine N, Jenkins GJ. High-throughput, multispecies, parallelized plasma stability assay for the determination and characterization of antibody–drug conjugate aggregation and drug release. ACS Omega. 2017 Aug 31;2(8):4207–15. doi:https://doi.org/10.1021/acsomega.7b00452.
- Bessire AJ, Ballard TE, Charati M, Cohen J, Green M, Lam MH, Loganzo F, Nolting B, Pierce B, Puthenveetil S. Determination of antibody–drug conjugate released payload species using directed in vitro assays and mass spectrometric interrogation. Bioconjug Chem. 2016 Jul 20;27(7):1645–54. doi:https://doi.org/10.1021/acs.bioconjchem.6b00192.
- Kabat EA, Wu TT, Perry HM, Foeller C, Gottesman KS. The nomenclature used to depict the mAb attachment sites follows that Described. In: Sequences of proteins of immunological interest. Darby (PA): Diane Publishing; 1992. ISBN 094137565X.
- Vester B, Rasmussen K. High performance liquid chromatography method for rapid and accurate determination of homocysteine in plasma and serum. Eur J Clin Chem Clin Biochem. 1991 Sep;29(9):549–54. doi:https://doi.org/10.1515/cclm.1991.29.9.549.
- Stabler SP, Marcell PD, Podell ER, Allen RH. Quantitation of total homocysteine, total cysteine, and methionine in normal serum and urine using capillary gas chromatography-mass spectrometry. Anal Biochem. 1987 Apr;162(1):185–96. doi:https://doi.org/10.1016/0003-2697(87)90026-1.
- Andersson A, Isaksson A, Hultberg B. Homocysteine export from erythrocytes and its implication for plasma sampling. Clin Chem. 1992 Jul;38(7):1311–15. doi:https://doi.org/10.1093/clinchem/38.7.1311.
- Salazar JF, Herbeth B, Siest G, Leroy P. Stability of blood homocysteine and other thiols: EDTA or acidic citrate? Clin Chem. 1999 Nov;45(11):2016–19.
- Rossi R, Milzani A, Dalle-Donne I, Giustarini D, Lusini L, Colombo R, Di Simplicio P. Blood glutathione disulfide: in vivo factor or in vitro artifact? Clin Chem. 2002 May;48(5):742–53.
- Oddoze C, Lombard E, Portugal H. Stability study of 81 analytes in human whole blood, in serum and in plasma. Clin Biochem. 2012 Apr;45(6):464–69. doi:https://doi.org/10.1016/j.clinbiochem.2012.01.012.
- Powis G, DeGraw CL. N-oxide reduction by hemoglobin, cytochrome C and ferrous ions. Res Commun Chem Pathol Pharmacol. 1980 Oct;30(1):143–50.
- Lavrijsen K, van Dyck D, van Houdt J, Hendrickx J, Monbaliu J, Woestenborghs R, Meuldermans W, Heykants J. Reduction of the prodrug loperamide oxide to its active drug loperamide in the gut of rats, dogs, and humans. Drug Metab Dispos. 1995 Mar;23(3):354–62.
- Kitamura S, Terada A, Inoue N, Kamio H, Ohta S, Tatsumi K. Quinone-dependent tertiary amine N-oxide reduction in rat blood. Biol Pharm Bull. 1998 Dec;21(12):1344–47. doi:https://doi.org/10.1248/bpb.21.1344.
- Kitamura S, Takekawa K, Sugihara K, Ohta S. Nonenzymatic reduction of N-hydroxy-2- acetylaminofluorene to 2-acetylaminofluorene by heme in the presence of hydroquinones. J Health Sci. 2000;46(1):66–69. doi:https://doi.org/10.1248/jhs.46.66.
- Sugihara K, Kitamura S, Ohta S, Tatsumi K. Reduction of hydroxamic acids to the corresponding amides catalyzed by rabbit blood. Xenobiotica. 2000 May;30(5):457–67. doi:https://doi.org/10.1080/004982500237479.
- Dorywalska M, Strop P, Melton-Witt JA, Hasa-Moreno A, Farias SE, Galindo Casas M, Delaria K, Lui V, Poulsen K, Sutton J, et al. Site-dependent degradation of a non-cleavable auristatin-based linker-payload in rodent plasma and its effect on ADC efficacy. PLoS One. 2015 Jul 10;10(7):e0132282. doi:https://doi.org/10.1371/journal.pone.0132282.
- Dorywalska M, Strop P, Melton-Witt JA, Hasa-Moreno A, Farias SE, Galindo Casas M, Delaria K, Lui V, Poulsen K, Loo C, et al. Effect of attachment site on stability of cleavable antibody drug conjugates. Bioconjug Chem. 2015 Apr 15;26(4):650–59. doi:https://doi.org/10.1021/bc5005747.
- Bahar FG, Ohura K, Ogihara T, Imai TJ. Species difference of esterase expression and hydrolase activity in plasma. J Pharm Sci. 2012 Oct;101(10):3979–88. doi:https://doi.org/10.1002/jps.23258.