
ABSTRACT
Biotherapeutics are exposed to common transition metal ions such as Cu(II) and Fe(II) during manufacturing processes and storage. IgG1 biotherapeutics are vulnerable to reactive oxygen species (ROS) generated via metal-catalyzed oxidation reactions. Exposure to these metal ions can lead to potential changes to structure and function, ultimately influencing efficacy, potency, and potential immunogenicity of the molecules. Here, we stress four biotherapeutics of the IgG1 subclass (trastuzumab, trastuzumab emtansine, anti-NaPi2b, and anti-NaPi2b-vc-MMAE) with two common pharmaceutically relevant metal-induced oxidizing systems, Cu(II)/ ascorbic acid and Fe(II)/ H2O2, and evaluated oxidation, size distribution, carbonylation, Fc effector functions, antibody-dependent cellular cytotoxicity (ADCC) activity, cell anti-proliferation and autophaghic flux. Our study demonstrates that the extent of oxidation was metal ion-dependent and site-specific, leading to decreased FcγRIIIa and FcRn receptor binding and subsequently potentially reduced bioactivity, though antigen binding was not affected to a great extent. In general, the monoclonal antibody (mAb) and corresponding antibody-drug conjugate (ADC) showed similar impacts to product quality when exposed to the same metal ion, either Cu(II) or Fe(II). Our study clearly demonstrates that transition metal ion binding to therapeutic IgG1 mAbs and ADCs is not random and that oxidation products show unique structural and functional ramifications. A critical outcome from this study is our highlighting of key process parameters, route of degradation, especially oxidation (metal catalyzed or via ROS), on the CH1 and Fc region of full-length mAbs and ADCs.
Abbreviations: DNPH 2,4-dinitrophenylhydrazine; ADC Antibody drug conjugate; ADCC Antibody-dependent cellular cytotoxicity; CDR Complementary determining region; DTT Dithiothreitol; HMWF high molecular weight form; LC-MS Liquid chromatography–mass spectrometry; LMWF low molecular weight forms; MOA Mechanism of action; MCO Metal-catalyzed oxidation; MetO Methionine sulfoxide; mAbs Monoclonal antibodies; MyBPC Myosin binding protein C; ROS Reactive oxygen species; SEC Size exclusion chromatography
Introduction
Large molecule biotherapeutics such as monoclonal antibodies (mAbs) and antibody-drug conjugates (ADCs) are important drug treatments for a wide range of diseases. Due to their potency and specificity to the target, these biologics can show remarkable efficacy compared to synthetic small molecule drugs. Citation1 However, unlike typical small molecule drugs, biotherapeutics are susceptible to physicochemical degradation that may lead to inactivation of the drug Citation2 and are especially susceptible to oxidation of several amino acids such as methionine (Met) and (Trp). Citation3,Citation4 These structural changes can substantially affect the quality, safety, and efficacy profile of biotherapeutics.
Oxidation of proteins occurs under a variety of physiological conditions. In nature, oxidation regulates cellular signaling Citation5 and neutrophilic antimicrobial activity, Citation6 but more damaging effects caused by reactive oxygen species (ROS) accumulation and associated with aging and several neurodegenerative diseases. Citation7 Oxidized proteins, such as enzymes, become highly sensitive to proteolytic degradation by proteosomes and can be rendered catalytically less active or inactive and more thermolabile.Citation8,Citation9 Therapeutic mAb oxidation can occur in the complementary-determining regions (CDRs), which recognize targeted antigens, and on the Fc of an antibody, which includes binding sites for the neonatal FcRn receptor, FcγRs, or complement C1q. Citation10 Modification in these regions can lead to loss of function, reduced binding affinity between IgG1 and its receptors, or produce structural changes that result in neo-epitopes that may raise immunogenicity concerns. Citation11
Metal-catalyzed oxidation (MCO) is one of the most widely studied and clinically relevant forms of oxidative stress-induced protein oxidation due to its irreversible nature. Citation12 Cu(II) and Fe(II/ III), two common catalytic redox-active transition metal ions, can be found in fermentation media, buffers/ excipients, and surfactant, or as trace contaminants from leaching of metal ions from fermentation vessels or container closure systems during storage and handling. Citation13,Citation14 Under redox conditions, both these metal ions can produce ROS and can lead to significant oxidation of IgG1 mAbs. Citation15–18 ROS generated during metal-catalyzed oxidation can also introduce carbonyl groups (reactive aldehydes or ketones), a widely used measure for oxidative stress, in the side chain of amino acids or the polypeptide backbone itself through cleavage of peptide bond by α- amidation pathway. Citation19,Citation20 Carbonylation functions as an internal protein quality control mechanism and is associated with aging, senescence, cancer, and aggregates found in Parkinson’s and Alzheimer’s disease. Citation9,Citation21 Recently, Aryal et al. Citation22 reported doxorubicin-mediated carbonylation of cardiac myosin binding protein C (MyBPC) in the presence of Fe(II/III). In this model, carbonylated MyBPC serves as a mechanistic indicator for loss of MyBPC activity and doxorubicin-induced cardiotoxicity and exemplifies the importance of investigating the biological impact of a variety of oxidized biotherapeutics.
Autophagy has been implicated in cell survival in HER2-overexpressing breast cancer cells and in developing resistance to trastuzumab. Citation23 Autophagy is a catabolic process that helps in recycling cellular components, including oxidized proteins. Citation24 Higher amounts of ROS have been found in tumor cells as opposed to the surrounding tissue. Citation25 These high levels of ROS could result in additional oxidative degradation of therapeutic proteins and removal by autophagy, as previously reported. Citation26,Citation27 However, a clear correlation between oxidized proteins and autophagy has not been reported for the IgG1 subclass of mAbs. While Fc receptor-dependent mechanisms contribute substantially to the action of cytotoxic antibodies against tumors in vivo,Citation28 whether the oxidation of amino acids in the Fc receptor-binding regions contributes to trastuzumab biological activity or resistance remain unclear.
We have previously reported analytical methods, structural changes and the functional consequences of metal binding and metal-catalyzed reactions toward mAbs. Citation29–31 Recently, we demonstrated site-specific binding of Cu(II) to the hinge region of the IgG1 subclass of mAbs. Citation29 This study was designed to reveal whether metal ions (that are either co-purified with the mAbs during upstream processes or interact with the mAbs under normal storage conditions) could initiate oxidation reactions under physiologically relevant redox conditions. We performed extensive and orthogonal characterization to understand the impact of MCO on physicochemical changes (e.g., oxidation, carbonylation, size variants) and on in vitro and cell-based activity (e.g., FcRn, FcγRIII binding, HER2 antigen binding, cell anti-proliferation, antibody-dependent cellular cytotoxicity (ADCC) activity, autophagy) of 4 different IgG1 biotherapeutics (trastuzumab, anti-NaPi2b and their corresponding drug conjugates trastuzumab emtansine/ T-DM1 and anti-NaPi2b-vc-MMAE, respectively). MCOs were based on previously optimized conditions. Citation32–34 Peptide-level characterization was obtained to provide further insight into the carbonylation, structural modifications and degradation of these biotherapeutics, with particular attention to the regions around metal ion binding.
Our findings demonstrate there is a correlation between the type of metal ion and changes in size variants and carbonyl formation. Overall, Cu(II)-mediated MCO (using Cu2+/ascorbic acid) has a greater effect on formation of low molecular weight forms (LMWF) and carbonyls despite the similar amino acid oxidation levels on the Fc region compared to Fe(II)-mediated MCO (using the Fe2+/H2O2 system). These oxidation products have an observable impact on FcγRIII and FcRn binding while not affecting HER2 binding or autophagic flux, suggesting selective degradation of the IgG1 sequence based on the type of metal exposure and its potential binding pockets. This work clarifies our understanding of the biological consequences of MCO-mediated mAb and ADC degradation and will add to the quality and potential safety repertoire of this important class of drug molecules.
Results
Structural and physicochemical effects of metal-catalyzed oxidation on biotherapeutics
M255 and M431 oxidation detected by LC-MS peptide map
Met 255 (M255) in the CH2 domain and Met 431 (M431) in the CH3 domain are established oxidation hotspots in IgGs. Citation35 We investigated the level of protein oxidation after treatment with either Cu(II)/ascorbic acid or Fe(II)/H2O2 by liquid chromatography-mass spectrometry (LC-MS) tryptic peptide mapping and used oxidation of M255 and M431 as the first measure of overall oxidation. These residues in trastuzumab, T-DM1, anti-NaPi2b, and anti-NaPi2b-vc-MMAE show the +16 Da mass shift indicative of the conversion of Met to methionine sulfoxide (MetO) after Cu(II) or Fe(II) treatment (). Cu(II) oxidized trastuzumab, T-DM1, anti-NaPi2b, and anti-NaPi2b-vc-MMAE all showed an increase in oxidation of M255 and M431 that ranged from ~19 to 26% and 6 to 12%, respectively, as compared to the control, 3.0 to 3.7% and 1.1 to 1.9%, respectively, after 24 h at 37°C ().
Figure 1. Effects of metal catalyzed oxidation (MCO) by Cu(II)/Asc and Fe(II)/H2O2 on Fc (a) M255 and (b) M431 by LC-MS Tryptic Peptide Mapping.

Similarly, oxidation by Fe(II) Fenton reaction showed a marked increase in oxidation at concentrations of 0.05 mM Fe(II)/1 mM H2O2 and 0.5 mM Fe(II)/0.1 mM H2O2. Lower Fe(II) (and higher H2O2) showed greater change after 3 hours at 37°C, indicating a greater impact on M255 and M431 oxidation with increasing amounts of H2O2 as compared to increasing Fe(II) concentration (). The increase in oxidation ranged from ~43 to 45% at M255 and ~24 to 27% oxidation at M431 for the 0.05 mM Fe(II)/1 mM H2O2 and 0.5 mM Fe(II)/ 0.1 mM H2O2, respectively, as compared to the control.
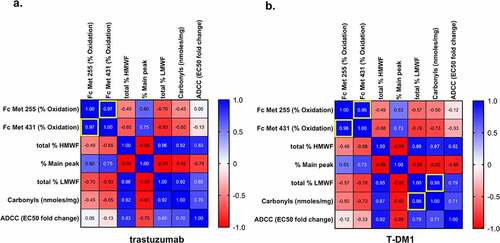
Pearson’s correlation analysis showed a positive correlation between Fc M255 and M431 oxidation for trastuzumab (r = 0.970, P = 0.030, 95% CI 0.1367 to 0.9994), T-DM1 (r = 0.960, P = 0.040, 95% CI −0.009 to 0.9992), and anti-NaPi2b (r = 0.961, P = 0.039, 95% CI −0.0035 to 0.9992) (). This indicates that changes in oxidation of the two sites are likely to follow the same trend due to either Cu(II)- and Fe(II)-mediated MCO. Anti-NaPi2b-vc-MMAE (r = 0.922, P = 0.078, 95% CI −0.3418 to 0.9984) shows a weaker correlation than anti-NaPi2b, its mAb counterpart, but showed similar increase in oxidation overall ().
Figure 2. Effects of MCO on (a) trastuzumab and (b) T-DM1 using Pearson correlation analysis. Pearson’s r-values outlined in yellow have P-value <0.05.
Effects of metal-catalyzed oxidation on total carbonylation by ELISA
Protein carbonylation is a direct result of MCO. Citation33,Citation36 Protein carbonylation can be determined by selective derivatization of carbonyl groups with 2,4-dinitrophenylhydrazine (DNPH) and measurement of the derivatized 2,4-dinitrophenylhydrazone (DNP) group in proteins using anti-DNP antibody. Spectrophotometry, ELISA, and western blot are commonly used analytical methods to determine carbonylation levels in proteins, with the more recent addition of Lucifer yellow carbohydrazide as a derivatization agent using size exclusion chromatography (SEC) analysis. Citation37
Both Cu(II) and Fe(II)-catalyzed oxidation showed an increase in carbonyls for all mAbs and ADCs tested (, ). However, Cu(II)-catalyzed oxidation significantly increased the total level of carbonylation of all four biotherapeutics, ranging from ~ 57 to 133-fold change. Formation of carbonyls after Cu(II) treatment appears to plateau after 3 hours using 0.01 mM Cu(II) and 1 mM ascorbate; however, at 1 mM Cu(II) and 4 mM ascorbic acid, a time-dependent increase in carbonylation was observed for trastuzumab, T-DM1 and anti-NaPi2b. A 28–63-fold increase in carbonylation was observed in the Fe(II)-mediated MCO in the presence of 1 mM H2O2 as compared to the changes observed with lower amounts of H2O2, which ranged from around 7- to 11-fold increase in total carbonylation. The results show that the higher amount of carbonyl correlates with increasing amounts of H2O2, and not with increasing amounts of Fe(II). When subjected to the same treatment condition, all four biotherapeutics showed similar total carbonyls. The presence of higher levels of carbonylation under Cu(II)-catalyzed oxidation as compared to Fe(II) was further confirmed by tryptic peptide analysis ().
Table 1. Impact of MCO on Fc Met255 and Met431 oxidation, size variants changes, and carbonyl formation for mAbs and ADCs after various time points at 37°C. n = 3.
Figure 3. Effects of MCO on carbonylation (nmols carbonyls/mg protein) for (a) trastuzumab, (b) T-DM1, (c) NaPi2b, and (d) NaPi2b-vc-MMAE after various time points at 37°C. The cross (+) indicates results of the untreated control and values that are not significantly different from the untreated control. Error bars represent standard deviation where n = 3. *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001.
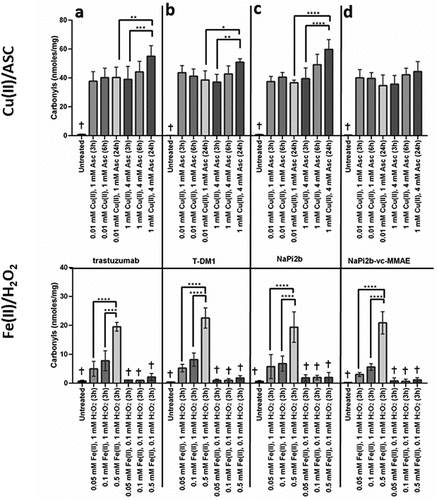
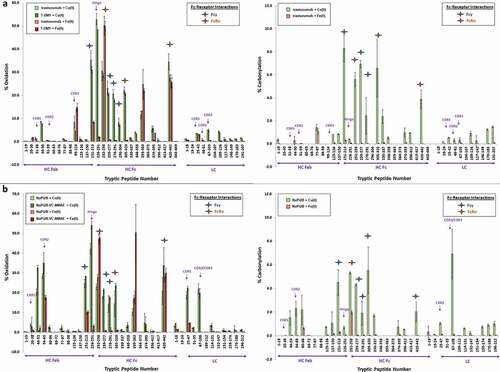
Figure 4. Impact of MCO on mAb and ADC peptide level (a, c) % oxidation and (b, d) % carbonylation as detected by LC-MS Tryptic Peptide Mapping. Treatment conditions = 0.01 mM Cu(II)/1 mM Asc 24 hr, 37°C and 0.05 Fe(II)/1 mM H2O2 up to 3hr, 37°C.
Effects of metal-catalyzed oxidation by size exclusion chromatography
Overall, the proteins that were Cu(II) treated showed major changes in aggregation, i.e., aggregates, or high molecular weight forms (HMWFs), and fragmentation, or LMWFs, as compared to Fe(II) (). For the trastuzumab and T-DM1 pair (mAb and ADC, respectively), a decrease in main peak with a significant concomitant increase in the HMWF (18 and 24%, respectively) and LMWF (25 and 20%, respectively) was observed after 6 hours at 37°C. Thereafter, the decrease in the main peak and increase in HMWF and LMWF continues at a slower rate up to 24 hours at 37°C in the presence of Cu(II) and ascorbic acid. The effect of Cu(II)-mediated oxidation on anti-NaPi2b and anti-NaPi2b-vc-MMAE pairs trend similarly to trastuzumab and T-DM1 ().
In contrast to Cu(II), trastuzumab and TDM1 oxidized with Fe(II) at various concentrations showed much less change in HMWF, main peak, and LMWF. The chromatographic profile shows extensive degradation such that there was no resolution between the main peak, HMWF, and LMWF under Cu(II) stressed conditions. On the other hand, the Fe(II) oxidized trastuzumab and T-DM1 showed a distinguishable profile, with a distinct main peak similar to the starting material (Figure S1a and Figure S1b).
In addition to major differences in chromatographic profiles observed between Cu(II) and Fe(II)-catalyzed oxidation, there were distinct differences in the amount of size variants between trastuzumab and T-DM1, respectively. For trastuzumab and T-DM1, the overall trend for % HMWF (ranked from highest to lowest change) was (): Cu(II) T-DM1 > Cu(II) trastuzumab ≫ Fe(II) T-DM1 > Fe(II) trastuzumab. The trend in % fragments was: Cu(II) trastuzumab > Cu(II) T-DM1 ≫ Fe(II) trastuzumab > Fe(II) T-DM1.
For anti-NaPi2b and anti-NaPi2b-vc-MMAE, the trend for the % HMWF was similar to the trend observed for trastuzumab and T-DM1 (i.e., Cu(II) NaP2b-vc-MMAE > Cu(II) NaP2b ≫ Fe(II) NaP2b-vc-MMAE > Fe(II) NaP2b). However, the percentage of fragments in the Cu(II) oxidized anti-NaPi2b was the highest observed (i.e., Cu(II) NaP2b > Cu(II) NaP2b-vc-MMAE > Fe(II) NaP2b-vc-MMAE > Fe(II) NaP2b). Overall, both the ADCs (T-DM1 and anti-NaPi2b-vc-MMAE) contained higher amounts of aggregates compared to the mAbs, trastuzumab and anti-NaPi2b.
LC-MS Tryptic Peptide Map Oxidation and Carbonylation Analysis Using Hydroxyl Radical Footprinting Data Analysis Workflow
LC-MS peptide map data were analyzed to evaluate the peptide-level oxidation and carbonylation of the IgG1 antibody. From the tryptic peptides, we can determine the regions and residues most susceptible for modification.
From these data, the relative percent (%) oxidation and relative (%) carbonylation at each peptide were determined. The total carbonylation (Pro, Lys, Arg, and Thr) was determined using the ELISA method (see total carbonylation section earlier), while this orthogonal method to quantitatively approximate the relative percent carbonylation per peptide at two residues Arg and Lys was also used. A few notable sites, i.e., the CDRs, hinge region, FcγRIIIa, FcRn binding sites, and their corresponding peptides were identified and analyzed further. The baseline level of oxidation was examined and determined to be <3% and <2% for M255 and M431 (), respectively, and overall very low carbonyl formation (<0.7 nmoles carbonyls/mg protein) (, ). The mAbs and their respective ADC showed generally similar profiles. However, the section prior to the hinge tryptic peptide 226–251, which includes the highly conserved upper hinge residues (VE219PKSCDK), Citation38 was undetected, as these sections were cut by trypsin into VEPK and SCDK in which the modified forms elute quickly. Citation39 Sequence coverage was obtained, but no hydroxy radical footprinting (HRF) analysis coverage was observed for these small early eluting peptides.
Using the HRF data analysis workflow, we observed that both Cu(II) and Fe(II) elicit extensive modification across the mAbs, albeit the extent and region-specific modifications were significantly metal-dependent. The trend in oxidation and carbonylation was similar for both mAb/ADC pairs, and therefore, here we focus in detail on the trastuzumab/T-DM1 pair. Overall, Cu(II) treatment produced significantly greater peptide level oxidation () and carbonylation () targeted in different regions than Fe(II) for trastuzumab and T-DM1, despite similar, or lower, levels of M255 and M431 oxidation. Anti-NaPi2b and NaPi2b-vc-MMAE also showed similar trends as trastuzumab (). Due to differing antigen binding residues, CDR2 for anti-NaPi2b and anti-NaPi2b-vc-MMAE contained more His oxidation than CDR3 in HC 99–124 (4.6–8.5%) and LC 67–103 (4.9–5.0%), Met-containing peptides for trastuzumab and T-DM1 under Cu(II) treatment. Additionally, little to no carbonylation in the CDR regions for anti-NaPi2b/ NaPi2b-vc-MMAE and trastuzumab/T-DM1 pairs was observed.
CDR antigen binding sites were also Cu(II) oxidized at low levels (< 3.6% in CDR1, < 0.7% in CDR2, and < 8.5% for CDR3) (). For Fe(II), we see similar low-level oxidation in CDR1 and CDR2 (<3.6%) and slightly higher amounts in CDR3 (< 15%), likely due to a Fe(II) susceptible oxidation site (). Little to no carbonylation on these same peptides were detected after Cu(II) and Fe(II)-catalyzed oxidation ().
Overwhelmingly, oxidation by Cu(II) was centered around the CH1-hinge-CH2 regions in the hinge peptide near peptides HC 151–213 and HC 226–251, with up to 35% and 53% oxidation, respectively, while Fe(II) showed significantly lower oxidation in the same region (up to 27% and 6.1%, respectively) (). Similarly, Cu(II) carbonylation on these same peptides was detected, with up to 8.3% and 0.4%, respectively, and with Fe(II) with little carbonyl formed (up to 1.3% and 1.7%, respectively) ().
Within the CH2 region HC 259–277 (21%-23%), HC 278–291 (18–21%), and HC 296–304 (7.3–8.3%) show significant oxidation due to Cu(II)-mediated MCO. Substantially lower levels were detected after Fe(II)-catalyzed oxidation, with 0.7–2.8%, 0.5–2.7%, and 0–0.6% for the three peptides, respectively. These peptides encompass the FcγRIIIa binding site. Likewise, there was also a noticeable increase in carbonylation, up to 6.9% in the Cu(II)-mediated MCO, while levels remain low (up to 0.5%) for all three HC peptides in the Fe(II)-mediated MCO.
The FcRn binding site peptides also exhibited significant oxidation, namely peptide HC 252–258 (29–31%), HC 305–320 (21–22%), HC 374–395 (5.4–5.8%), and HC 420–442 (28–35%) showed oxidation increase. Fe(II) showed increased oxidation, especially in the M255 and M431 containing peptide HC 252–258 (21–50%), HC 305–320 (0.5–1.9%), HC 374–395 (0.6–4.2%), and HC 420–442 (13–26%). Carbonylation due to Cu(II)-catalyzed oxidation resulted in 1.0–6.9%, whereas little to no carbonylation was detected with Fe(II).
Functional effects of metal-catalyzed oxidation on biotherapeutics
Surface plasmon resonance for HER2, FcγRIIIa and FcRn binding of trastuzumab
We selected trastuzumab to further investigate the structure-function relationship in a representative, well-studied mAb biotherapeutic. Surface plasmon resonance (SPR) can isolate the molecular interactions without the complex signal cascades of cell-based methods Citation40 (which was also used and described in subsequent sections). The SPR results () indicated that the Cu(II)-stressed molecules displayed the largest impact on FcγIIIa and FcRn binding. Under Cu(II) conditions, no response for FcγIIIa binding could be detected for both samples. Cu(II)-oxidized samples after 6 and 24 hours resulted in approximately 250- and 2500-fold reduction in FcRn binding, respectively, whereas Fe(II)-catalyzed oxidation displayed minor decreases (~ 2-fold) in both FcγIIIa and FcRn binding.
Table 2. Effects of MCO on trastuzumab binding to FcRn, FcγRIIIa, and HER2 antigen after metal catalyzed oxidation as measured by SPR.
Antigen binding to HER2 was also performed for the trastuzumab with Cu(II) and Fe(II) treatment. While there was a small decrease in the results of the binding affinity for both metals, the overall binding affinity for HER2 was largely maintained and not dramatically affected (). No differences in direct binding of the CDR region were observed between Cu(II) and Fe(II).
ADCC activity
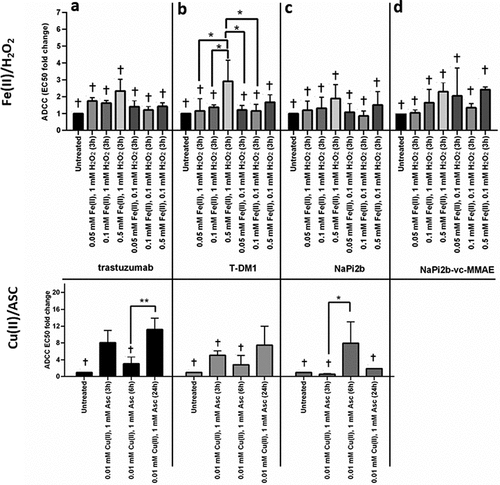
ADCC activity is an important mechanism of action for biological potency of mAbs and ADCs. Cu(II)- and Fe(II)-catalyzed oxidizing conditions led to decreased ADCC activity for trastuzumab, T-DM1, anti-NaPi2b and anti-NaPi2b-vc-MMAE (). Cu(II) oxidizing conditions had greater effects on ADCC activity, with significant decreases in ADCC activities (i.e., higher EC50 values) for trastuzumab (24 hours, 9-fold, P = 0.0002, 95% CI −15.79 to −4.580), T-DM1 (24 hours, 7-fold, P = 0.0181. 95% CI −12.07 to −0.8555), anti-NaPi2b (6 hours, 2-fold, P = 0.0243, 95% CI −13.22 to −0.6818). Anti-NaPi2b-vc-MMAE exceeded high signal, and it is not included in our statistical analysis or in . Fe(II) oxidized samples showed a decrease up to 2-fold for trastuzumab and T-DM1, no change for NaPi2b, and up to 3-fold for anti-NaPi2b-vc-MMAE. Notably, for Cu(II), the ADC NaPi2b –vc-MMAE showed high loss in ADCC activity, which exceeded the limits of the method, as compared to the corresponding mAb anti-NaPi2b, which showed < 8% loss. For the Fe(II) oxidation system, ADCC activity also decreased, up to approximately 1- to 3-fold, but was considerably less than observed with Cu(II) ().
Figure 5. Effects of MCO on Antibody-Dependent Cellular Cytotoxicity (ADCC) for (a) trastuzumab, (b) T-DM1, (c) NaPi2b, and (d) NaPi2b-vc-MMAE at 37°C after various timepoints. The cross (+) indicates results of the untreated control and values that are not significantly different from the untreated control. Error bars represent standard deviation where n = 3. *p < 0.05, **p < 0.01. Note that signal with anti-NaPi2b-vc-MMAE exceeded the upper limit of quantification and was therefore not included in panel d for statistical analyses.
Anti-proliferation bioassay
Trastuzumab was also chosen to further elucidate the structure-function impact of MCO using a cell-based anti-proliferation bioassay Citation41 that measures inhibition of the tumor cell line growth. Briefly, the anti-proliferation method offers us the ability to further elucidate the biological effects that are dependent on complex cellular cascade signals that SPR, i.e. binding constants, alone does not capture.Citation42 After 24 hours at 37°C, Cu(II) oxidation of trastuzumab showed no specific activity for anti-proliferation, as there appeared to be a complete loss of potency. In contrast, Fe(II) and H2O2 oxidation retained specific activity, but with ~5.6- fold decrease as compared to the trastuzumab control ().
Table 3. Effects of MCO on BT-474 cell proliferation for trastuzumab.
Autophagy
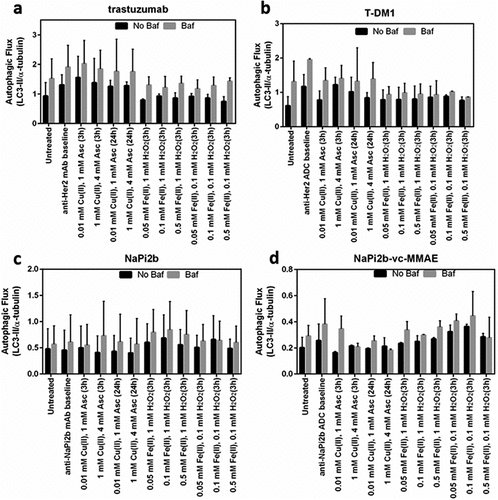
The ability of each IgG1 biotherapeutic to induce catabolic autophagy was assessed in cell lines expressing the cell surface antigen for each mAb or ADC drug. Autophagy was assessed in the presence and absence of bafilomycin (baf), which inhibits autophagic flux by preventing the fusion of autophagosomes and lysosomes, resulting in accumulation of autophagy-specific LC3-II. Both ADCs tested had cytotoxic activity against target cell lines at 20 µg/mL. Therefore, autophagic flux was assessed in target cells treated with 200 ng/mL of T-DM1 and anti-NaPi2b-vc-MMAE or 20 µg/mL of trastuzumab and antiNaPi2b. For all mAbs and ADCs tested in this model, neither oxidized nor unoxidized forms led to changes in autophagic flux compared to untreated cells (). No clear relationship between Cu- and Fe-mediated oxidation and autophagy was observed as none of the biotherapeutic drugs were strong inducers of autophagy.
Figure 6. Impact of MCO on autophaghy for (a) trastuzumab, (b) T-DM1, (c) NaPi2b, and (d) NaPi2b-vc-MMAE.
Discussion
Impact of metal-catalyzed oxidation on structure and function
Metal Ion binding and generation of reactive oxygen species
We have previously reported a site-specific binding pocket for the redox active Cu(II) ions on large therapeutic proteins. Binding and hydrolytic fragmentation was mediated by His residues, but also driven in part by geometric considerations and neighboring amino acids. Citation29 Similar to Cu(II), which has a particularly high affinity and predominantly binds to His as well as Asp and Glu (e.g., peptide NH2Gly-Gly-HisN-methylamide Kd = 2.07 x 10–17), Citation43 Fe(II/III) have similar binding affinity to Asp, Tyr, and His residues (Kd ~10−22 to 10–20 M in ferritin Citation44,Citation45). While there is limited information for the concentration of leachable copper during manufacturing process, leachable iron has been studied and it has been reported that leaching of iron is highly affected by pH, temperature, and formulation buffer.Citation46 It has also been reported that up to 172.15 µg/L (or 3.1 mM) of iron was extracted from rubber stopper in acidic condition (3% acetic acid) and the concentration was further increased up to 1393 µg/L in the presence of chelating agents such as diethylenetriaminepentaacetic acid (DTPA).Citation47 We anticipate that the oxidation reactions observed herein signify a stressed condition, but the degradation profile would still be representative of those observed if such contaminants /impurities were to be found in trace amounts in manufacturing, biologic formulations, storage or processing steps. Citation33,Citation46–48
We have also shown recently that redox inactive metal ions such as Zn(II) also can bind to a specific His residue in an IgG1 mAb and can lead to oligomerization. Citation31 All of these examples indicate and implicate His residues for site-specific metal ion binding to mAbs.
Unlike Zn(II), redox active metal ions such as Cu(II) and Fe(II/III) have the demonstrated potential to cause oxidative damage to therapeutic proteins.Citation49 In the presence of electron donating species such as ascorbic acid or electron acceptor hydrogen peroxide, redox active metals could lead to formation of localized ROS that could damage neighboring amino acids. The reaction center generates ROS such as ●OOH, and the extremely reactive ●OH. While the ●OH generated at the reaction center is a powerful oxidant, the reaction is fast and diffusion limited, i.e., the impact distance for ●HO is only a few nanometers, while that for H2O2 is approximately 1.5 mm. Citation50 Herein, we report a metal ion-specific impact on product quality with a clear trend of structure-function relationship to both the mAbs (trastuzumab and antiNaPi2b) and their corresponding ADCs (T-DM1 and anti-NaPi2b-vc-MMAE). Our study shows the metal-catalyzed reactions not only leads to oxidation, but changes aggregation, fragmentation, binding, and potency of these biotherapeutics.
Impact of metal-catalyzed oxidation to structural and physicochemical properties of biotherapeutics
The highly conserved and surface-exposed IgG1 Fc Met255 and Met431 are labile sites for both Cu(II)- and Fe(II)-mediated oxidation Citation34,Citation51 and a well-studied surrogate quality attribute to determine the extent of oxidative degradation of the biotherapeutics we evaluated. In this study, we observed that Fe(II) leads to oxidation of peptides that contain Met, while peptides that contain both His and Met or His alone are oxidized mainly by Cu(II) for all 4 biotherapeutics. Here, the oxidation ratio for Met255:Met431 was approximately ~1.1 to 3.0:1, which is consistent with the range reported in previous studies. Citation34,Citation51 The positive correlation shown between % oxidation of Fc Met255 and Fc Met 431 in trastuzumab, T-DM1, and anti-NaPi2b supports this finding. Percent oxidation of anti-NaPi2b-vc-MMAE was also consistent with the other molecules, although a statistical correlation was not evident. Altogether, these results indicate that oxidation of the two highly conserved Fc Met sites is likely to trend in the same direction, with the oxidizing treatments containing either Cu(II) or Fe(II), and support the metal was indeed effective in oxidizing the molecules. Met255 is relatively more surface exposed than Met431 and readily accessible for oxidative reactions. Citation52 Despite the limited surface exposure of Met431 and oxidation at a slower rate, either Cu(II) or Fe(II) can bind and react at nearby ligands and generate ROS that could oxidize both Met255 and Met431. Overall, our data indicate that Fe(II)-mediated MCO leads to higher Met oxidation specifically in the Fc region than Cu(II)-mediated MCO ().
Site specificity for oxidation reactions was more predominant when mediated by Cu(II) than by Fe(II). A region that is particularly prone to Cu(II) binding is the upper hinge of an IgG1. Our current results reveal that Cu(II)-mediated oxidative stress increases site-specific oxidation, as well as the other physicochemical attributes studied here, i.e., carbonylation and size variants, particularly fragmentation. Cu(II)-catalyzed oxidation has a particularly profound impact on the flexible hinge region (peptides HC 226–251) where we have previously proposed a Cu(II)-specific binding pocket. Citation29
Physicochemical changes occurred during both Cu(II) and Fe(II) exposure that increased carbonyl and size variants (both fragments and aggregates) and were observed under conditions that promoted extensive M255 and M431 oxidation. Fe(II) Fenton-based generation of ROS can potentially generate higher methionine oxidation (), but lower carbonylation and fragmentation than Cu(II) (). A positive correlation was seen with the MCO-induced total carbonyl and percent fragment formation for only T-DM1 (r = 0.977, p < 0.05) (). It is possible that an increase of carbonylation could lead to low molecular weight fragment formation via fragmentation as byproducts of MCO. Carbonylation can occur at the most susceptible residues (i.e., Pro, Lys, Arg, and Thr) that may lead to subsequent peptide bond cleavage (glutamyl, aspartyl, and prolyl side chains) and protein fragmentation through diamide and α-amidation pathways. Citation53
Additionally, increases in aggregation were observed under Cu(II)-catalyzed oxidation, resulting in more HMWF as compared to Fe(II)-catalyzed oxidation. Increases in carbonylation as a result of oxidation by Fe(II) (i.e., as stainless-steel leachate) has shown concomitant aggregation due to changes in hydrophobicity. Citation54–56 Radical-based protein oxidation has also been known to induce covalent aggregation, especially causing formation of disulfide and dityrosine cross-links, Citation57 thereby causing more hydrophobic patches that can induce aggregation. Citation55 Additionally, changes in structure from Met255 and Met431 oxidation can further destabilize the CH2-CH3 region to cause possible aggregation. Citation58
Differential product quality impact of mAbs versus ADCs
ADCs are created by conjugating cytotoxic agents either via a lysine (e.g., T-DM1) or cysteine (e.g., NaPi2b-vc-MMAE) in the mAbs. Conjugating a hydrophobic drug could affect various product qualities of the resulting molecule as demonstrated previously. Citation59,Citation60 However, specific product quality impacts due to oxidation reactions have not been reported previously. Our results demonstrate the nonspecific conjugation at the Cys and Lys residues for vc-MMAE and DM1 drug linkers, respectively, did not make the mAb more susceptible to chemical modifications, particularly for the hinge conjugation of vc-MMAE ( and ) and that the MCO affected both the mAb and the ADC similarly. For total protein carbonylation, there were no major differences or unique trends observed after Cu(II) or Fe(II) oxidation between trastuzumab and T-DM1. This trend was also consistent for the anti-NaPi2b and anti-NaPi2b-vc-MMAE mAb-ADC pair.
An inherent ADC property that was shown to also be perpetuated under MCO is the higher propensity of aggregate formation for T-DM1 and anti-NaPi2b-vc-MMAE, as they exhibited substantially more aggregates compared to their corresponding mAbs after MCO. Generally, with the added linker-drug the ADCs tend to be more hydrophobic and can promote self-association. Citation61 We hypothesize that the addition of additional oxygen to the molecule after MCO potentially makes them even more hydrophobic. Met oxidation was observed to directly cause increased aggregation during MCO, with MCO soluble aggregates Citation62 and HMWFs containing neoepitopes, such as from oxidation, shown to be immunogenic in transgenic mice studies. Citation63
Functional effects of metal-catalyzed oxidation on the mAb and ADC
The physicochemical structural changes caused by Cu(II)- and Fe(II/III)-mediated oxidation can have profound implications that lead to changes in function, i.e., ADCC activity, PK, and antiproliferation. For trastuzumab and T-DM1, the CDR residues bind to the overexpressed extracellular domain of HER2, which leads to activation of several mechanisms of action, including antiproliferation, growth inhibition, and apoptosis, that function directly on the tumor cells, as well as ADCC activity, which attracts immune cells to tumor sites that overexpress HER2. Citation64–67 ADCC activity is initiated upon antigen binding on the tumor by CDR receptors of the antibody followed by interaction with FcγRIIIa. There is also evidence that the antigen-binding fragment (Fab) may interact directly with the Fc effector function receptors. Citation68–71 NaPi2b and NaPi2b-vc-MMAE targets the sodium-dependent phosphate transporter of solid tumors, such as ovarian, non-squamous non-small cell lung, and papillary thyroid cancers, via binding in the CDR region. Citation72 Both ADCs carry a chemotherapeutic agent that targets tumor genesis once internalized into the cell. Citation73,Citation74 Although all four biotherapeutics possess similar Fc, the relative ADCC activity by NaPi2b and NaPi2-vc-MMAE as part of its clinical MOA needs further study since these antibodies target membrane-bound targets and can produce variable anti-tumor effects. Citation75,Citation76
Another prominent Fc receptor that can affect PK is the neonatal receptor FcRn, which is involved in serum half-life and cellular transport mechanisms. The binding of FcRn to Fc occurs at the interface of the CH2-CH3 domains where both Met255 and Met430 are found. Citation64–67,Citation77 His435 found in the IgG1 CH2-CH3 domain has also been implicated in FcRn binding. Citation78 Since Cu(II) coordination and oxidation is shown to have particularly high affinity toward His residues, we observed a significant impact to FcRn binding as well.
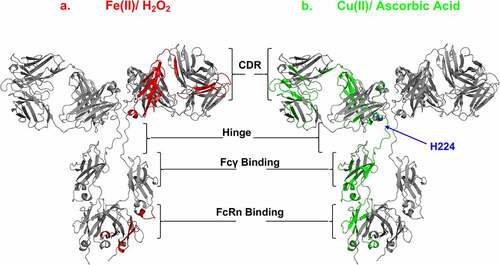
Cu(II)-catalyzed oxidation clearly affected the ADCC-mediated FcγRIIIa receptor binding residues in the lower hinge and CH2 regions more than Fe(II)-mediated oxidation of trastuzumab (), T-DM1, anti-NaPi2b, and anti-NaPi2b-vc-MMAE. In conjunction with the peptide oxidation analysis, our results identified the areas in which both Cu(II)- and Fe(II)-catalyzed oxidation appeared to overlap (the CDR in the VL-CL and FcRn binding region in the CH2-CH3 domains), which prominently contained labile Met and His residues as targeted sites for oxidation, as shown in the trastuzumab homology structure (). Given the binding pocket around hinge His220, Cu(II)-catalyzed oxidation specifically targets residues in the hinge and those involved in FcγRIIIa binding. Susceptible Cu(II) binding sites in the Fcγ region include H268, and neighboring residues such as D265, S267, E270, and D270. Examining peptide HC259-277 by HRF workflow analysis, higher oxidation is indeed shown under Cu(II)-catalyzed conditions compared to Fe(II) ().
Figure 7. Effects of MCO on the mAb and ADC CDR, hinge, FcRIII and FcRn binding sites. IgG1 homology structure with mapped oxidation levels impacted by either (a) Fe(II)/H2O2 (red) or (b) Cu(II)/Asc (green) to the functional sites. Note: hinge residue “H224” is oxidized specifically by Cu(II), and not Fe(II). Cu(II) Model of IgG1 constructed using 1BJ1 and 1IGY crystal structures
We observed biological effects of Cu(II)-catalyzed oxidation that trends with the other physicochemical results and show higher efficiency over Fe(II) despite similar levels of Fc Met-O. All four molecules oxidized by Fe(II) and Cu(II) showed decreasing ADCC activity (i.e., increasing EC50), which is consistent with the various levels of FcγRIIIa binding region oxidation. Fe(II)-catalyzed oxidation led to about a 3.5-fold lower ADCC activity among the four biotherapeutics tested here compared to their unoxidized controls. Similarly, Cu(II)-catalyzed oxidation for the two mAbs, trastuzumab and NaPi2b, showed significant decreases in ADCC activity, 9 to 2 –fold, respectively, while T-DM1 showed a 6.5-fold reduction. However, NaPi2b-vc-MMAE, the other ADC, showed virtually no ADCC response under Cu(II)-catalyzed oxidation conditions as compared to anti- NaPi2b, trastuzumab, and T-DM1, in spite of having similar levels of carbonylation.
The cell-based antiproliferation findings further align with the trend observed with Cu(II) and Fe(II) in SPR binding for the effector functions, as Cu(II)-catalyzed oxidation showed completely diminished activity as compared to the 5-fold decrease in antiproliferation by Fe(II). The complete loss in antiproliferation activity was observed despite partial oxidation to CDR3 (< 9%), which contains Met107, due to Cu(II)-catalyzed oxidation. Based on previous studies, Asn-30 (CDR1) deamidation, Asp102 (CDR3) isomerization, or Trp105 (CDR3) oxidation Citation79 can lead to an impact in potency, while Met107 is typically buried and not prone to oxidation. Citation52 Unlike Fe(II)-catalyzed oxidation, Cu(II)-catalyzed oxidation greatly affected the FcγRIIIa binding region; therefore, it is not surprising to see significantly diminished ADCC (engagement with effector cells), although a complete loss in antiproliferation activity of trastuzumab with Cu(II)-catalyzed oxidation () was unexpected.
Another important effector function, besides FcγRIIIa, is the FcRn binding site that is involved in serum half-life and cellular transport and located in the interface of the CH2-CH3 domains containing Met255 and Met431. Citation64–67,Citation77 A previous study showed that oxidation of IgG1 Met252 at 79% and Met428 at 57% impair its serum half-life. Citation35 Studies using Met site-directed mutagenesis as surrogates to actual oxidation show that Met252 causes more extensive deleterious FcRn binding effect than Met428. Citation51 In our studies, MCO-mediated Met oxidation showed differences in FcRn binding based on the type of metal ion used. The amount of Fe(II) induced FcRn binding by SPR showed a 2-fold decrease for trastuzumab. However, Cu(II) showed 2500 – fold reduction in FcRn binding, indicating not only that Met residues are important to binding to FcRn, but also that ROS-mediated degradation around the metal ion binding sites leads to other product quality impacts (such as carbonylation, fragmentation or aggregation) and these can inactivate the overall FcRn binding.
Cu(II) and Fe(II) oxidation seem to affect different Fc effector function regions due to the specificity of the metal to the amino acids in the receptor binding region of the mAbs. This potentially leads to more rapid IgG degradation and shortened serum half-life, in the case of FcRn, or loss in ADCC activity for FcγRIIIa, as seen for Fe(II)- and Cu(II)-mediated MCO, respectively.
The SPR results indicate that MCO did not lead to any significant reduction in HER2 binding. Interestingly, the results from the cell-based antiproliferation bioassay showed both Cu(II) and Fe(II) impacted biological activity significantly, with the complete elimination of tumor-killing activity after Cu(II) MCO. In contrast, previous studies demonstrated trastuzumab containing as high as 50% M255 oxidation by iron did not lead to potency loss as determined by receptor binding assay and the same antiproliferative cell assay.Citation15
In addition to antiproliferation and ADCC activity as the primary and secondary mechanisms of action, respectively, for trastuzumab and other biotherapeutics, autophagy, also known as programmed cell death type II, is a complex cellular process that has been implicated as an alternative pathway of tumor cell death. Autophagy functions by preventing the potential to form tumors,Citation80 as well as a mechanism of cellular resistance by promoting tumorigenesis and eliminating drug from cancer cells.Citation23
These studies showed that in the presence of metabolic and hypoxic stresses, autophagic responses initiate protective mechanisms to respond to changing conditions to recognize and degrade proteins and organelles in periods of starvation. Citation81,Citation82 In the case of trastuzumab, acquired resistance to the drug was observed through the increase in LC3 structures (marker for autophagy) when HER2-overexpressing SKBR2 breast cancer cells were subjected to incremental exposure to trastuzumab (> 10 months), causing desensitization to the drug as a mechanism of cell survival. Citation23 A similar study was also performed for T-DM1 that corroborated the results in gastric cancer. Citation83 Additionally, inhibition of autophagic pathways was shown to be an effective target for cancer biotherapies, especially after prolonged treatment and exposure to the drug therapies.
In this study, cancer cells treated with trastuzumab, T-DM1, anti-NaPi2b, and anti-NaPi2b-vc-MMAE that clearly underwent oxidation catalyzed by Cu(II) and Fe(II) with the production of ROS did not show any meaningful differences in autophagy flux as compared to the control. Previous studies that subjected the cells to prolonged exposure to trastuzumab and T-DM1 showed that autophagy-induced drug resistance was a potential mechanism of tumor cell survival. Our study was unable to show clear evidence that autophagy could be initiated after oxidative degradation of biotherapeutics under the experimental conditions tested.
Statistical and correlation analyses
According to the two-way ANOVA analyses, the oxidation of M255 and M431 showed significant statistical interaction between the drug and treatment variables, indicating that the effect of treatment depends on the drug. Similar interaction is observed for the SEC-HPLC data. Carbonylation in Cu(II) treatments and ADCC in iron treatments do not have statistical interaction (P = 0.1661) and the drugs carbonylation and ADCC activity, respectively, are independently affected by the drug and the treatment. In Fe(II) oxidation, treatment conditions (P < 0.0001) drove the carbonylation similarly in all drugs tested. ADCC data in Cu(II) treated samples showed statistical interaction between the drug and treatment variables (P = 0.0041), indicating that the effect of treatment depends on the drug.
The Pearson’s correlations between the variables studied rely on the mean values in each treatment condition for each variable. Since both Cu(II) - and Fe(II)-containing conditions were included in the analysis, the correlations observed reflect changes in variables in response to any used treatment condition. The Met255 and Met431 % oxidation correlation for trastuzumab, T-DM1, and anti-NaPi2b shows significance even with the limited amount of data available for analysis. The T-DM1 is the only drug studied that had positive correlation between carbonylation and total low molecular weight species (P = 0.023, 95% CI 0.2506 to 0.9995) as treatment conditions were varied.
Key study results
In our study, Cu(II)/ascorbic acid oxidation system clearly surpassed Fe(II)/H2O2 in the effectiveness of oxidation and results in region-specific structural and functional impact on trastuzumab, T-DM1, anti-NaPi2b, and anti-NaPi2b-vc-MMAE. Our results demonstrate that MCO causes definitive physicochemical and structural changes in biotherapeutics as shown by changes in oxidation, carbonylation, and size variants. While the peptide-level oxidation (i.e., CDR) and size-variant changes depend on the drug studied, the change in carbonylation depends on the oxidization conditions. These changes affected critical Fc effector function binding regions that drive ADCC and resulted in diminished potency, notably more so by Cu(II) than Fe(II/III). The findings clearly highlight the higher risk of copper as an available trace metal in biotherapeutic drug products.
One of the most interesting of our observations is the complete elimination of the antiproliferative effects of trastuzumab after Cu(II)-mediated oxidation. While 9–10% CDR oxidation does not seem to affect HER2 binding, overall physicochemical changes that include fragmentation, carbonylation and aggregation affected antiproliferation activity of breast cancer cells. These were unexpected results given there was minimal CDR modifications and no significant loss in antigen bindings. Moreover, if the loss in antiproliferation was due to complete loss in antigen binding, it should have been reflected in the ADCC data. Instead, the loss in ADCC mirrored the loss in FcγRIIIa binding, suggesting there was still full antigen engagement. These results are also in agreement with those reported by Carter et al. Citation84 wherein the antiproliferative activity of several humAb4D5 variants against p185HERCitation2-overexpressing SK-BR-3 cells did not correlate with their binding affinity for p185HERCitation2 extracellular domain.
The mechanism by which trastuzumab elicits an antiproliferative response in HER2-overexpressing cells is thought to be from one of three potential pathways (reviewed in Hudis et al. Citation85): 1) blocking of the cleavage of the HER2 extracellular domain, 2) sterically hindering of homo- and hetero- receptor dimerization, or 3) receptor degradation through internalization. Although there are significant amounts of Cu(II)-induced aggregation and fragmentation under the conditions tested, it could not account for the complete loss in activity. Based on these data, the loss of antiproliferation is likely a mAb-mediated (e.g., metal-catalyzed higher order structural effects in the monomer of trastuzumab), rather than a target cell-based response. However, the exact nature of the structure-function relationship remains unknown and requires additional studies with engineered mAbs lacking domains of interest.
Our data clearly indicate that MCO leads to a loss of Fc receptor-mediated mechanisms, including loss of ADCC. Resistance to trastuzumab treatment has been shown through the induction of autophagy, Citation23,Citation86 but other literature suggests the induction of autophagy improves HER2-treatments, Citation87 including T-DM1.Citation88 In this study we found no measurable differences in autophagy, indicating that metal ion-mediated oxidation of these biotherapeutics did not induce this pathway under the conditions tested, and therefore is not likely a prominent mechanism for eliminating oxidized proteins or rescuing the cells.
Spiridon et al. Citation89 reported that the in vitro antiproliferative activity of both full-length IgG and its F(ab’)2 fragments was similar. However, they found that IgG had significant antitumor activity in vivo whereas the F(ab’)2 fragments were only marginally effective even at 5x doses, showing the importance of the Fc-mediated interactions for antitumor activity. In addition, Sun et al. recently demonstrated that Fab-FcγRIIIa interactions mediate ADCC activity. Citation71 While the contribution of the Fab interaction to ADCC may not be as strong as the Fc region or glycan interactions, the current work shows that Cu(II)-induced significant MCO within the same region of the identified Fab-FcγRIIIa interaction. Whether a combination of loss of Fc-mediated functions could lead to trastuzumab resistance is still unclear, but our MCO-mediated structure-function work reported here indicates that immune-mediated mechanisms to destroy tumor cells are critical, and any oxidizing mechanisms that could impede this activity require careful scrutiny. Finally, the regional and site-specific oxidation we observed corroborates a metal binding pocket for Cu(II) in the upper hinge region of IgG1 subclass of mAbs, as well as the significant impact to various biological activities of this class of biotherapeutics.
Since MCO did not lead to reduced antigen binding due to low amounts of CDR oxidation, but led to reduced Fc receptor-mediated biological activity, one critical outcome from our study is the revelation of the importance of process parameters and important quality attributes, especially metal catalyzed oxidation, on the CH1 and Fc region of full-length mAbs.
We have previously reported oxidative stress under normal storage conditions for drug products as a result of light exposure.Citation90 While the reaction conditions (e.g., photomediated versus metal ion-mediated) are different, the resultant oxidation to amino acids such as Met and Trp is due to common reactive oxygen species, such as singlet oxygen, hydroxyl radicals and hydrogen peroxide. Mechanistically speaking these reactive oxygen species play a significant role in the long-term stability of mAbs and ADCs. Oxidation, either by light- or metal ion-mediated reactions, could result in altered potency or purity and have other important biological consequences such as immunogenicity. Interestingly photo-Fenton reactions have also been reported that indicate that reactive oxygen species generated by light could interact with trace metal ions such as Fe(III) to induce a Fenton-type oxidation reaction.Citation91 Such mechanisms could adversely affect the stability of liquid protein formulations.
Materials and methods
Materials
All IgG1 mAbs and ADCs were manufactured by Genentech to study the impact of metal-catalyzed oxidation. Trastuzumab, T-DM1, anti-NaPi2b, and anti-NaPi2b-vc-MMAE pairs have the same antibody molecule and the ADC is the same mAb conjugated with a cytotoxic agent. All mAbs and ADCs were formulated at 8 mg/mL in a formulation buffer containing 10 mM sodium acetate, trihydrate (JT Baker cat# 3461), 60 mM sucrose (Ferro Pfanstiehl, cat #S-124-1), 0.002% polysorbate 20 (JT-Baker cat 4112–04), pH 5.5. The Cu(II)SO4 (cat #C8027), Fe(II)SO4 (cat #215422), L-ascorbic acid (cat #A7506-100 g), hydrogen peroxide 30% (w/w) in H2O containing stabilizer (cat #H1009), DTPA (cat #D1133), EDTA (cat # E9884), were purchased from Sigma-Aldrich and Methionine (cat #30415) from Ajinomoto. The centriprep centrifugal filter unit, 15 mL 10 kDa cutoff was purchased from Millipore (cat #4304).
mAb/ADC metal oxidation conditions
To prepare the Cu(II) oxidation conditions, solutions were prepared to contain ~3 mg/ mL mAb/ADC concentration in 10 mM sodium acetate, 60 mM sucrose, 0.02 mg/mL PS20, pH 5.5 with 0.01 mM Cu(II)SO4 + 1 mM ascorbic acid or 1 mM Cu(II)SO4 + 4 mM ascorbic acid and stored at 37°C up to 6 and 24 hours. The reaction was quenched by addition of Met and DTPA to 1 mM. For the iron stress conditions, solutions were prepared to contain ~3 mg/ mL mAb/ADC concentration in 10 mM sodium acetate, 60 mM sucrose, 0.02 mg/mL PS20, pH 5.5 with 0.05 mM Fe(II)SO4 + 1 mM H2O2, 0.5 mM Fe(II) SO4 + 0.1 mM H2O2, or 0.1 mM Fe(II)SO4 + 1 mM H2O2 and stored at 37°C up to 1.5 and 3 hours. The reaction was quenched by addition of Met and DTPA to 1 mM. The metal-treated samples were buffer exchanged into formulation buffer with centripreps.
Three independent experimental solutions were prepared, unless otherwise indicated, and tested using the analytical methods as indicated. Protein concentration and pH were measured for each sample to confirm target concentration and pH. Samples were filtered through 0.22 micron filter, aliquoted, and stored at 2–8°C or frozen if not used immediately.
Analytical methods
Size exclusion chromatography
Size variant distributions of the mAbs were determined by SEC using a TosoHaas Bioscience column G3000 SWXL (South San Francisco, CA) with an autosampler set at 5°C on an Agilent 1200 HPLC (Santa Clara, CA). All samples were injected at 50-μg load onto the column and eluted over 28 minutes with 0.2 M potassium phosphate, 0.25 M potassium chloride, 5% isopropyl alcohol (pH 6.9) mobile phase at a flow rate of 0.5 mL/min. Protein elution was monitored at 280 nm. Results are reported as relative percent, which is the area of an individual peak divided by the total area under the curve. Data analysis was performed on all chromatograms and integration was done using Thermo Scientific Chromeleon software (Sunnyvale, CA).
Liquid chromatography-mass spectrometry tryptic peptide mapping
Oxidation of protein peptides was monitored using a tryptic peptide digest followed by LC-MS analysis. Samples of mAbs were prepared for LC-MS analysis as follows. Proteins were denatured by diluting 165 μg of each sample to 1.2 mg/mL with a reduction and carboxymethylation buffer (6 M guanidine HCl, 320 mM Tris, and 2 mM EDTA, pH 7.0). Following denaturation, dithiothreitol (DTT) was added (17 mM) and the samples were incubated at 37°C for 1 hour to reduce the proteins. The samples were then carboxymethylated by the addition of iodoacetic acid in 1 N NaOH to a concentration of 40 mM iodoacetic acid, then stored in the dark at room temperature for 15 min. The alkylation reaction was quenched by the addition of DTT to a final concentration of 8 mM. The reduced and alkylated samples were buffer exchanged (PD MultiTrap™ G-25 plate; GE Healthcare) into trypsin digestion buffer (100 mM Tris, 2 mM CaCl2, pH 7.5). Sequencing-grade trypsin was added at an enzyme-to-protein ratio of 1:20 by weight to digest the samples. The digestion reaction was incubated at 37°C for 1 hour and then quenched by adding 54 mM L-methionine in 80% formic acid (FA) to the sample to a final FA concentration of 3%.
Peptide mapping was performed on a Waters Acquity I-Class UHPLC coupled to a Thermo Q Exactive Plus mass spectrometer. Separation of a 10-μL protein injection was performed on an Acquity UPLC Peptide CSH C18 column (130 Å, 1.7 µm, 2.1 mm x 150 mm) with the column temperature controlled at 77°C. Solvent A consisted of 0.1% FA in water and solvent B consisted of 0.1% FA in acetonitrile.
Data were processed using Thermo Chromeleon™ software. Data were analyzed by integrating extracted ion chromatograms of the monoisotopic m/z using the (+1) and (+2) charge states for the native tryptic peptide and the oxidized tryptic peptide of Fc Met255 and the (+3), (+4) and (+5) charge states for the native tryptic peptide and the oxidized tryptic peptide of FcMet431.
Note that all amino acids and peptides are sequentially numbered.
A 10-ppm error tolerance was used. The relative percentage of oxidation was calculated by dividing the peak area of the oxidized peptide species by the sum of the peak area of the native and oxidized peptides. Only methionine sulfoxide (+16 Da) was used to calculate methionine oxidation as methionine sulfone (Mþ32) was not observed under these conditions.
Hydroxyl radical footprinting data analysis workflow using protein metrics software suite analysis
For analysis of the acquired LC-MS Peptide Map data, Protein Metrics software Byonic was used to identify the fragmentation data for each peptide sample, and percent oxidation was calculated using the extracted mass of each identified species from the precursor scans (350–2000 m/z) with Biologic, as previously described by Lin et al. and Garcia et al. Byonics search parameters include: carboxy methyl, oxidation, dioxidation, Trp conversion to kynurenin, custom modification for oxidation, and carbonylation.Citation92
Determination of protein carbonylation by ELISA assay
Selected mAbs/ADCs were diluted to a final concentration of 1 mg/mL in 10 mM sodium acetate, 60 mM sucrose, 0.02 mg/mL PS20, pH 5.5 and treated with 1) Cu(II)SO4 to a final concentration of 0.01 mM and 1 mM, and ascorbate to a final concentration of 1 and 4 mM; or 2) Fe(II)SO4 to a final concentration of 0.05, 0.1 and 0.5 mM in presence of H2O2 to a final concentration of 0.1 and 1 as indicated in . After incubation for 3, 6, or 24 hours at 37°C, metal-catalyzed redox reactions were stopped using 1 mM DTPA and immediately processed for quantification of carbonyl modifications using a modified ELISA method as described in the previous publications.Citation25,Citation93 Briefly, 0.5–1 µg of each sample was diluted to 10 µL and mixed with an equal volume (10 µL) of 10% (w/v) sodium dodecyl sulfate (SDS). The samples were then derivatized with DNP using 20 µL of 20 mM DNPH solution prepared in 10% (v/v) trifluoroacetic acid. After incubation at room temperature for 10 minutes with occasional vortexing every 2 minutes, the reaction was neutralized with 20 µL of 2 M Tris base. A 3 µL aliquot of DNP-derivatized sample was diluted with 0.25 mL of adsorption buffer (20 mM NaHCO3, 150 mM NaCl, 0.25% SDS (w/v), pH 8.5), and 100 µL of diluted samples were loaded in duplicate on to a 96-well Maxisorp plate (Thermo Fisher Scientific, Rockford, IL). The plate was covered with aluminum foil and incubated overnight at 4°C. After incubation, the sample wells in a 96-well plate were rinsed gently 6 times with PBST (1X phosphate-buffered saline (PBS) containing 0.05% Tween 20) and incubated with 200 µL of blocking buffer (1% bovine serum albumin (BSA) in PBST) for 1 hour at 37°C. After removal of blocking buffer, plate was incubated with 100 µL of blocking buffer containing goat anti-DNP antibody (ThermoFisher, catalog # A150-117A) in each well for 1 hour at room temperature. Following incubation, the plate was rinsed 6 times with PBST, and incubated with horseradish peroxidase (HRP)-conjugated rabbit anti-goat IgG antibody (Agilent, catalog # P044901-2) for 1 hour at room temperature. After washing 6 times with PBST, the plate was incubated with 100 µL of TMB substrate per well at room temperature for 2–3 minutes for color development. The reaction was stopped with 100 µL of 0.5 M H2SO4 and the absorbance was measured at 450 nm and 690 nm. After subtraction of background absorbance at 690 nm, the carbonyl content in each sample was determined using a standard curve generated from an oxidized BSA standard in the same plate. Carbonyls are reported as nmoles carbonyls/ mg protein.
Cell culture
SK-Br-3 cells (target for anti-HER2 antibody drugs), and OVCAR-3 cells (target for anti-NaPi2b antibody drugs) were obtained from ATCC. MDA-MB-231 cells were cultured in DMEM/F-12 (Mediatech) containing 10% fetal bovine serum (FBS), 2 mM L-glutamine, 1 mM sodium pyruvate, and penicillin/streptomycin. SK-Br-3 cells were maintained in McCoy’s media (Gibco) supplemented with 10% FBS and penicillin/streptomycin. OVCAR-3 cells were maintained in RPMI 1640 (Corning) media supplemented with 20% FBS, 2 mM L-glutamine, 0.01 mg/ml bovine insulin, and penicillin/streptomycin. ADCC effector cells were purchased from Promega and cultured in RPMI-1640 medium supplemented with 10% FBS 2 mM L-glutamine, and 1 mM sodium pyruvate, 100 µg/mL hygromycin, 250 µg/mL antibiotic G-418 sulfate, and 0.1 mM MEM non-essential amino acids. All cells were cultured at 37°C in a humidified 5% CO2 atmosphere.
ADCC assay
ADCC activity was measured using the Promega ADCC Reporter Bioassay following the manufacturer’s instructions. Serial dilutions of oxidized and non-oxidized antibody drugs were prepared before applying to target cell lines. For trastuzumab, T-DM1, anti-NaPi2b, and anti-NaPi2b-vc-MMAE, serial dilutions gave a range of 1 µg/mL to 15.2 pg/mL final concentrations. Effector cells expressing FcγRIIIa and an NFAT response element driving expression of luciferase were then added. Assays were performed in 96-well plates with an effector to target ratio of 150,000:30,000 cells/well for trastuzumab and anti-NaPi2b targeting drugs. Co-cultures were incubated for 18 hours at 37°C. Luciferase activity was then measured using Bio-Glo Luciferase Assay System (Promega) and luminescence was measured on a SpectraMax i3 microplate reader (Molecular Devices). Data were analyzed as fold-induction of luminescence compared to no antibody control and EC50 was determined by fitting log10([antibody, μg/mL]) versus response using GraphPad Prism. Data are reported as least three independent experiments.
Autophagy assays
To measure autophagic flux, changes in LC3 protein level were measured in target cells treated with oxidized or non-oxidized antibody drugs in the presence and absence of Bafilomycin A1 (Baf). Target cells (SK-BR-3 for trastuzumab/T-DM1 and OVCAR3 for anti-NaPi2b/ anti-NaPi2b-vc-MMAE) were plated and allowed to adhere to culture dishes for 24 hours before treatment with oxidized or non-oxidized drug (20 µg/mL for trastuzumab and anti-NaPi2b; 200 ng/mL for T-DM1 and anti-NaPi2b-vc-MMAE) for 24 hours. For indicated cultures, 5 nM bafilomycin was added for the last 2 hours of culture. Cells were collected and lysed using RIPA with protease and phosphatase inhibitors. Cell lysates were then analyzed by western blotting using the primary antibodies, anti-LC3B (Novus Biologicals, NB100-2220) anti-α-tubulin (Cell Signaling, 2144), and anti-rabbit secondary antibody (LI-COR). Images were collected and densitometry analyzed using an Odyssey imager (LI-COR). Autophagic flux was calculated by dividing the LC3-II by the α-tubulin signal. For each treatment, results for are shown for treatment with and without bafilomycin. Each experiment was performed twice.
SPR binding assay
All SPR measurements were done on a Biacore T200 instrument (GE Healthcare). FcRn, FcγIIIa and HER2 were immobilized on a CM5 chip (GE Healthcare) through amine coupling using manufacturer recommended conditions. The reference channel (Fc1) was prepared by conducting a blank immobilization. PBS pH 7.4 was used as running buffer and 10 mM Glycine pH 2.0 was used as regeneration buffer for FcγIIIa and antigen binding experiments. 10 mM sodium phosphate, 150 mM sodium chloride, pH 6.0 was used as running buffer and PBS pH 7.4 was used as regeneration buffer for FcRn binding experiments. Datasets that showed binding were reference subtracted and fit to a 1:1 binding model using the BIAevaluation software (GE Healthcare).
BT-474 cell proliferation assay
BT-474 human breast carcinoma cells (ATCC HTB 20) were grown in DMEM:F12 (1:1) medium (Gibco) supplemented with 10% FBS (HyClone) and 2 mM glutamine (Gibco). BT-474 cells were harvested and cell suspensions of 1.0 × 105 cells per-ml were made; 100 μl were dispensed per-well in BD Falcon 96-well tissue culture plates followed by preincubating for 1–3 hours at 37°C. A total of 100 μL of samples and standards of varying concentrations were added to the assay plate and incubated for 5 days at 37°C. At the end of the incubation, the viability was quantitated by adding 25 μl of AlamarBlue™ (Invitrogen) per well. AlamarBlue™ is a redox dye that fluoresces when reduced by live cells. Following addition of AlamarBlue™, assay plates were incubated for 6–8 hours at 37°C to allow time for cells to metabolize dye. At the end of the incubation, assay plates were cooled for 10 minutes. Relative fluorescence (530 nm excitation, 590 nm emission) was measured using a Spectramax M5e plate reader. A 4-parameter logistic curve-fitting program was used to generate a standard curve that reports the trastuzumab concentration. Specific activity was relative to the anti-proliferative activity of the reference lot (assigned 101% specific activity). Results reported as normalized values compared to the trastuzumab control.
Statistical and correlation analyses
Data analyses were performed by Excel, and GraphPad Prism.
2-Way ANOVA
2-way ANOVA with Bonferroni’s multiple comparison’s tests were performed on ADCC and Carbonylation data using alpha-level of 0.05. The ADCC data were analyzed as fold changes to accommodate the assumption of normality required for 2-way ANOVA. The data used in 2-way ANOVA analyses followed approximate normal distribution. The anti-NaPi2b-vc-MMAE ADCC data were not included in the analysis because it had no activity detected to allow for statistical analysis.
Pearson correlation coefficient
Correlation analysis using Pearson correlation was performed on four treatment conditions: Cu(II)SO4 0.01 mM with Asc 1 mM 6 hours, CuSO4 0.01 mM with Asc 1 mM 24 hours, Fe(II)SO4 0.05 mM in 1 mM H2O2 3 hours, and iron 0.5 mM in 0.1 mM H2O2 3 hours. All values used were averaged from three replicates for the analysis. Pearson’s r and p-values were used to find likely correlations between the different variables included in the analysis: Fc Met255 (% Oxidation), Fc Met431 (% Oxidation), total % HMWF, % Main peak, total % LMWF, Carbonyls (nmoles/mg), and ADCC (EC50 fold change). The total % HMWF, % Main peak, total % LMWF correlations with each other were not considered significant as they are interdependent. Each drug was analyzed separately.
Supplemental Material
Download TIFF Image (116.7 MB)Acknowledgments
The authors would like to thank Emily Holz (Genentech), Drs. Maria Teresa Gutierrez-Lugo (FDA), Susan Kirshner (FDA) and Tongzhong Ju (FDA) for critical reading of the manuscript.
Disclosure statement
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the U.S. Food and Drug Administration and the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2122957
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Chinthaginjala H, Kalpana K, Manchikanti SP, Reddy PG, Karamthoy S, Vennapusa NR. Biotherapeutics as drugs, its delivery routes and importance of novel carriers in biotherapeutics. Int J Of Pharma Sci Res. 2021;12:44–19.
- Makurvet FD. Biologics vs. small molecules: drug costs and patient access. Med. Drug Discovery. 2021;9:100075. doi:10.1016/j.medidd.2020.100075.
- Ji JA, Zhang B, Cheng W, Wang YJ. Methionine, tryptophan, and histidine oxidation in a model protein, PTH: mechanisms and stabilization. J Pharm Sci. 2009;98(12):4485–500. doi:10.1002/jps.21746.
- Sreedhara A, Lau K, Li C, Hosken B, Macchi F, Zhan D, Shen A, Steinmann D, Schoneich C, Lentz Y. Role of surface exposed tryptophan as substrate generators for the antibody catalyzed water oxidation pathway. Mol Pharm. 2013;10(1):278–88. doi:10.1021/mp300418r.
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82(1):47–95. doi:10.1152/physrev.00018.2001.
- Oliver CN. Inactivation of enzymes and oxidative modification of proteins by stimulated neutrophils. Arch Biochem Biophys. 1987;253(1):62–72. doi:10.1016/0003-9861(87)90637-0.
- Stadtman ER. Metal ion-catalyzed oxidation of proteins: biochemical mechanism and biological consequences. Free Radic Biol Med. 1990;9(4):315–25. doi:10.1016/0891-5849(90)90006-5.
- Stadtman ER, Oliver CN. Metal-catalyzed oxidation of proteins. Physiological consequences. J Biol Chem. 1991;266(4):2005–08. doi:10.1016/S0021-9258(18)52199-2.
- Levine RL. Carbonyl modified proteins in cellular regulation, aging, and disease. Free Radic Biol Med. 2002;32(9):790–96. doi:10.1016/S0891-5849(02)00765-7.
- Chen Y, Doud E, Stone T, Xin L, Hong W, Li Y. Rapid global characterization of immunoglobulin G1 following oxidative stress. MAbs. 2019;11(6):1089–100. doi:10.1080/19420862.2019.1625676.
- Boll B, Bessa J, Folzer E, Rios Quiroz A, Schmidt R, Bulau P, Finkler C, Mahler HC, Huwyler J, Iglesias A, et al. Extensive chemical modifications in the primary protein structure of Igg1 subvisible particles are necessary for breaking immune tolerance. Mol Pharm. 2017;14(4):1292–99. doi:10.1021/acs.molpharmaceut.6b00816.
- Stadtman ER, Levine R. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2004;25:207–18.
- Munir NN, Sangita C, Vikram S. Implications of trace levels of redox-active metals in drug-product formulation. BioPharm International. 2014;27(4).
- Graham RJ, Bhatia H, Yoon S. Consequences of trace metal variability and supplementation on Chinese hamster ovary (CHO) cell culture performance: a review of key mechanisms and considerations. Biotechnol Bioeng. 2019;116(12):3446–56. doi:10.1002/bit.27140.
- Lam XM, Yang JY, Cleland JL. Antioxidants for prevention of methionine oxidation in recombinant monoclonal antibody HER2. J Pharm Sci. 1997;86(11):1250–55. doi:10.1021/js970143s.
- Kohen R, Nyska A. Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol Pathol. 2002;30(6):620–50. doi:10.1080/01926230290166724.
- Torosantucci R, Schoneich C, Jiskoot W. Oxidation of therapeutic proteins and peptides: structural and biological consequences. Pharm Res. 2014;31(3):541–53. doi:10.1007/s11095-013-1199-9.
- Nassar M, Chandrasekharan S, Sadineni V. Implications of trace levels of redox-active metals in drug-product formulation. BioPharm International. 2014;27(4).
- Levine RL, Williams JA, Stadtman ER, Shacter E. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 1994;233:346–57.
- Stadtman ER. Covalent modification reactions are marking steps in protein turnover. Biochemistry. 1990;29(27):6323–31. doi:10.1021/bi00479a001.
- Nyström T. Role of oxidative carbonylation in protein quality control and senescence. Embo J. 2005;24(7):1311–17. doi:10.1038/sj.emboj.7600599.
- Aryal B, Jeong J, Rao VA. Doxorubicin-induced carbonylation and degradation of cardiac myosin binding protein C promote cardiotoxicity. Proc Natl Acad Sci U S A. 2014;111(5):2011–16. doi:10.1073/pnas.1321783111.
- Vazquez-Martin A, Oliveras-Ferraros C, Menendez J, Najbauer J. Autophagy facilitates the development of breast cancer resistance to the Anti-HER2 monoclonal antibody trastuzumab. PLoS ONE. 2009;4(7):e6251. doi:10.1371/journal.pone.0006251.
- Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22(3):377–88. doi:10.1038/cdd.2014.150.
- Aryal B, Rao VA, Ahmad A. Specific protein carbonylation in human breast cancer tissue compared to adjacent healthy epithelial tissue. PLoS One. 2018;13(3):e0194164. doi:10.1371/journal.pone.0194164.
- Kaushik S, Cuervo AM. Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol Aspects Med. 2006;27(5–6):444–54. doi:10.1016/j.mam.2006.08.007.
- Xiong Y, Contento AL, Nguyen PQ, Bassham DC. Degradation of oxidized proteins by autophagy during oxidative stress in Arabidopsis. Plant Physiol. 2007;143(1):291–99. doi:10.1104/pp.106.092106.
- Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6(4):443–46. doi:10.1038/74704.
- Glover ZK, Basa L, Moore B, Laurence JS, Sreedhara A. Metal ion interactions with mAbs: part 1. MAbs. 2015;7(5):901–11. doi:10.1080/19420862.2015.1062193.
- Rathnayaka H, Mozziconacci O, Sreedhara A, Schoneich C. Fragmentation of a monoclonal antibody by peroxotungstate. Pharm Res. 2018;35(11):219. doi:10.1007/s11095-018-2496-0.
- Mehta S, Flores H, Walters B, Sreedhara A. Metal ion interactions with mabs: part 2. zinc-mediated aggregation of IgG1 monoclonal antibodies. Pharm Res. 2021;38(8):1387–95. doi:10.1007/s11095-021-03089-7.
- Grewal P, Mallaney M, Lau K, Sreedhara A. Screening methods to identify indole derivatives that protect against reactive oxygen species induced tryptophan oxidation in proteins. Mol Pharm. 2014;11(4):1259–72. doi:10.1021/mp4007375.
- Kryndushkin D, Rao VA. Comparative effects of metal-catalyzed oxidizing systems on carbonylation and integrity of therapeutic proteins. Pharm Res. 2016;33(2):526–39. doi:10.1007/s11095-015-1807-y.
- Narhi LO, Luo Q, Wypych J, Torosantucci R, Hawe A, Fujimori K, Nashed-Samuel Y, Jawa V, Joubert MK, Jiskoot W. Chemical and biophysical characteristics of monoclonal antibody solutions containing aggregates formed during metal catalyzed oxidation. Pharm Res. 2017;34(12):2817–28. doi:10.1007/s11095-017-2262-8.
- Wang W, Vlasak J, Li Y, Pristatsky P, Fang Y, Pittman T, Roman J, Wang Y, Prueksaritanont T, Ionescu R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol Immunol. 2011;48(6–7):860–66. doi:10.1016/j.molimm.2010.12.009.
- Shacter E. Quantification and significance of protein oxidation in biological samples. Drug Metab Rev. 2000;32(3–4):307–26. doi:10.1081/DMR-100102336.
- Yang Y, Mah A, Yuk IH, Grewal PS, Pynn A, Cole W, Gao D, Zhang F, Chen J, Gennaro L, et al. Investigation of metal-catalyzed antibody carbonylation with an improved protein carbonylation assay. J Pharm Sci. 2018;107(10):2570–80. doi:10.1016/j.xphs.2018.06.015.
- Jacobsen FW, Padaki R, Morris AE, Aldrich TL, Armitage RJ, Allen MJ, Lavallee JC, Arora T. Molecular and functional characterization of cynomolgus monkey IgG Subclasses. Eur. J. Immunol. 2011;186(1):341–49. doi:10.4049/jimmunol.1001685.
- Vlasak J, Ionescu R. Fragmentation of monoclonal antibodies. mAbs. 2011;3(3):253–63. doi:10.4161/mabs.3.3.15608.
- Schasfoort RBM, Abali F, Stojanovic I, Vidarsson G, Terstappen LWMM. Trends in SPR cytometry: advances in label-free detection of cell parameters. Biosensors. 2018;8(4):102. doi:10.3390/bios8040102.
- Harris RJ, Kabakoff B, Macchi FD, Shen FJ, Kwong M, Andya JD, Shire SJ, Bjork N, Totpal K, Chen AB. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J Chromatogr B Biomed Sci Appl. 2001;752(2):233–45. doi:10.1016/S0378-4347(00)00548-X.
- Kelley RF, O’Connell MP, Carter P, Presta L, Eigenbrot C, Covarrubias M, Snedecor B, Bourell JH, Vetterlein D. Antigen binding thermodynamics and antiproliferative effects of chimeric and humanized anti-p185HER2 antibody Fab fragments. Biochemistry. 1992;31(24):5434–41. doi:10.1021/bi00139a003.
- Regan L. The design of metal-binding sites in proteins. Annu Rev Biophys Biomol Struct. 1993;22:257–87. doi:10.1146/annurev.bb.22.060193.001353.
- Baker HM, Anderson BF, Baker EN. Dealing with iron: common structural principles in proteins that transport iron and heme. Proc Natl Acad Sci U S A. 2003;100(7):3579–83. doi:10.1073/pnas.0637295100.
- Winter WE, Bazydlo LA, Harris NS. The molecular biology of human iron metabolism. Lab Med. 2014;45(2):92–102. doi:10.1309/LMF28S2GIMXNWHMM.
- Zhou S, Schöneich C, Singh SK. Biologics formulation factors affecting metal leachables from stainless steel. AAPS PharmSciTech. 2011;12(1):411–21. doi:10.1208/s12249-011-9592-3.
- Li X, Qian P. Identification of an exposure risk to heavy metals from pharmaceutical-grade rubber stoppers. J. Food Drug Anal. 2017;25(3):723–30. doi:10.1016/j.jfda.2016.07.008.
- Fliszar KA, Walker D, Allain L. Profiling of metal ions leached from pharmaceutical packaging materials. PDA J. Pharm. Sci. Technol. 2006;60:337–42.
- Mehta S, Flores H, Walters B, Sreedhara A. Metal ion interactions with mabs: Part 2. Zinc-mediated aggregation of IgG1 monoclonal antibodies. Pharm Res. 2021;38:1387–95.
- Davies Michael J. Protein oxidation and peroxidation. Biochemical Journal. 2016;473(7):805–25. doi:10.1042/BJ20151227.
- Gao X, Ji JA, Veeravalli K, Wang YJ, Zhang T, McGreevy W, Zheng K, Kelley RF, Laird MW, Liu J, et al. Effect of individual Fc methionine oxidation on FcRn binding: met252 oxidation impairs FcRn binding more profoundly than Met428 oxidation. J Pharm Sci. 2015;104(2):368–77. doi:10.1002/jps.24136.
- Shen FJ, Kwong M, Keck RG, Harris RJ. The application of tert-butylhydroperoxide oxidation to study sites of potential methionine oxidation in a recombinant antibody. Techniques in Protein Chemistry. 1996;7:275–84.
- Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272(33):20313–16. doi:10.1074/jbc.272.33.20313.
- Bee JS, Davis M, Freund E, Carpenter JF, Randolph TW. Aggregation of a monoclonal antibody induced by adsorption to stainless steel. Biotechnol Bioeng. 2010;105(1):121–29. doi:10.1002/bit.22525.
- Uehara H, Rao VA. Metal-mediated protein oxidation: applications of a modified ELISA-based carbonyl detection assay for complex proteins. Pharm Res. 2015;32(2):691–701. doi:10.1007/s11095-014-1496-y.
- Yang Y, Mah A, Yuk IH, Grewal PS, Pynn A, Cole W, Gao D, Zhang F, Chen J, Gennaro L, et al. Investigation of metal-catalyzed antibody carbonylation with an improved protein carbonylation assay. J Pharm Sci. 2018;107(10):2570–80.
- Zheng K, Ren D, Wang YJ, Lilyestrom W, Scherer T, Hong JKY, Ji JA. Monoclonal antibody aggregation associated with free radical induced oxidation. Int J Mol Sci. 2021;22(8):3952. doi:10.3390/ijms22083952.
- Burkitt W, Domann P, O’Connor G. Conformational changes in oxidatively stressed monoclonal antibodies studied by hydrogen exchange mass spectrometry. Protein Sci. 2010;19(4):826–35. doi:10.1002/pro.362.
- Beckley NS, Lazzareschi KP, Chih H-W, Sharma VK, Flores HL. Investigation into temperature-induced aggregation of an antibody drug conjugate. Bioconjug Chem. 2013;24(10):1674–83. doi:10.1021/bc400182x.
- Adem YT, Schwarz KA, Duenas E, Patapoff TW, Galush WJ, Esue O. Auristatin antibody drug conjugate physical instability and the role of drug payload. Bioconjug Chem. 2014;25(4):656–64. doi:10.1021/bc400439x.
- Acchione M, Kwon H, Jochheim CM, Atkins WM. Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody-drug conjugates. MAbs. 2012;4(3):362–72. doi:10.4161/mabs.19449.
- Joubert MK, Luo Q, Nashed-Samuel Y, Wypych J, Narhi LO. Classification and characterization of therapeutic antibody aggregates. J Biol Chem. 2011;286(28):25118–33. doi:10.1074/jbc.M110.160457.
- Boll B, Bessa J, Folzer E, Ríos Quiroz A, Schmidt R, Bulau P, Finkler C, Mahler HC, Huwyler J, Iglesias A, et al. Extensive chemical modifications in the primary protein structure of Igg1 subvisible particles are necessary for breaking immune tolerance. Mol Pharm. 2017;14(4):1292–99.
- Pohlmann PR, Mayer IA, Mernaugh R. Resistance to Trastuzumab in Breast Cancer. Clinical Cancer Research: an Official Journal of the American Association for Cancer Research. 2009;15(24):7479–91. doi:10.1158/1078-0432.CCR-09-0636.
- Junttila TT, Parsons K, Olsson C, Lu Y, Xin Y, Theriault J, Crocker L, Pabonan O, Baginski T, Meng G, et al. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res. 2010;70(11):4481–89. doi:10.1158/0008-5472.CAN-09-3704.
- Vu T, Claret FX. Trastuzumab: updated mechanisms of action and resistance in breast cancer. Front Oncol. 2012;2:62. doi:10.3389/fonc.2012.00062.
- Nakajima T, Okayama H, Ashizawa M, Noda M, Aoto K, Saito M, Monma T, Ohki S, Shibata M, Takenoshita S, et al. Augmentation of antibody-dependent cellular cytotoxicity with defucosylated monoclonal antibodies in patients with GI-tract cancer. Oncol Lett. 2018;15(2):2604–10. doi:10.3892/ol.2017.7556.
- Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10(5):301–16. doi:10.1038/nri2761.
- Jiang X-R, Song A, Bergelson S, Arroll T, Parekh B, May K, Chung S, Strouse R, Mire-Sluis A, Schenerman M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov. 2011;10(2):101–11. doi:10.1038/nrd3365.
- Yogo R, Yamaguchi Y, Watanabe H, Yagi H, Satoh T, Nakanishi M, Onitsuka M, Omasa T, Shimada M, Maruno T, et al. The Fab portion of immunoglobulin G contributes to its binding to Fcγ receptor III. Sci Rep. 2019;9(1):11957. doi:10.1038/s41598-019-48323-w.
- Sun Y, Izadi S, Callahan M, Deperalta G, Wecksler AT. Antibody–receptor interactions mediate antibody-dependent cellular cytotoxicity. Int. J. Biol. Chem.International. 2021;297(1):100826. doi:10.1016/j.jbc.2021.100826.
- Banerjee S, Oza AM, Birrer MJ, Hamilton EP, Hasan J, Leary A, Moore KN, Mackowiak-Matejczyk B, Pikiel J, Ray-Coquard I, et al. Anti-NaPi2b antibody-drug conjugate lifastuzumab vedotin (DNIB0600A) compared with pegylated liposomal doxorubicin in patients with platinum-resistant ovarian cancer in a randomized, open-label, phase II study. Annals of Oncology: Official Journal of the European Society for Medical Oncology. 2018;29(4):917–23. doi:10.1093/annonc/mdy023.
- Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011;11(2):263–75. doi:10.1586/era.10.226.
- Cumnock K, Tully T, Cornell C, Hutchinson M, Gorrell J, Skidmore K, Chen Y, Jacobson F. Trisulfide modification impacts the reduction step in antibody-drug conjugation process. Bioconjug Chem. 2013;24(7):1154–60. doi:10.1021/bc4000299.
- Peters C, Brown S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep. 2015;35(4). doi:10.1042/BSR20150089.
- Yu J, Song Y, Tian W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J Hematol Oncol. 2020;13(1):45. doi:10.1186/s13045-020-00876-4.
- Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. 2018;9(1):63–73. doi:10.1007/s13238-017-0473-8.
- Kim JK, Firan M, Radu CG, Kim CH, Ghetie V, Ward ES. Mapping the site on human IgG for binding of the MHC class I-related receptor, FcRn. Eur J Immunol. 1999;29(9):2819–25. doi:10.1002/(SICI)1521-4141(199909)29:09<2819::AID-IMMU2819>3.0.CO;2-6.
- Hensel M, Steurer R, Fichtl J, Elger C, Wedekind F, Petzold A, Schlothauer T, Molhoj M, Reusch D, Bulau P. Identification of potential sites for tryptophan oxidation in recombinant antibodies using tert-butylhydroperoxide and quantitative LC-MS. PLoS One. 2011;6(3):e17708. doi:10.1371/journal.pone.0017708.
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–76. doi:10.1038/45257.
- Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65(8):3336–46. doi:10.1158/0008-5472.CAN-04-3640.
- Cao X, Hu X, Zhang X, Gao S, Ding C, Feng Y, Bao W, Permyakov EA. Identification of metal ion binding sites based on amino acid sequences. PLOS ONE. 2017;12(8):e0183756. doi:10.1371/journal.pone.0183756.
- Zhang J, Fan J, Zeng X, Nie M, Chen W, Wang Y, Luan J, Zhu Z, Chang X, Ju D, et al. Targeting the autophagy promoted antitumor effect of T-DM1 on HER2-positive gastric cancer. Cell Death Dis. 2021;12(4):288. doi:10.1038/s41419-020-03349-1.
- Carter P, Presta L, Gorman CM, Ridgway J, Henner D, Wong W, Rowland AM, Kotts C, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proceedings of the National Academy of Sciences. 1992;89(10):4285–89. doi:10.1073/pnas.89.10.4285.
- Hudis CA. Trastuzumab–mechanism of action and use in clinical practice. N Engl J Med. 2007;357(1):39–51. doi:10.1056/NEJMra043186.
- Rodríguez-Muela N, Hernández-Pinto AM, Serrano-Puebla A, García-Ledo L, Latorre SH, de la Rosa Ej, Boya P, de la Rosa EJ. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 2015;22(3):476–87. doi:10.1038/cdd.2014.203.
- Janser FA, Tschan MP, Langer R. The role of autophagy in HER2-targeted therapy. Swiss Med Wkly. 2019;149:w20138. doi:10.4414/smw.2019.20138.
- Liu P, Fan J, Wang Z, Zai W, Song P, Li Y, Ju D. The role of autophagy in the cytotoxicity induced by trastuzumab emtansine (T-DM1) in HER2-positive breast cancer cells. AMB Express. 2020;10(1):107. doi:10.1186/s13568-020-01044-0.
- Spiridon CI, Guinn S, Vitetta ES. A comparison of the in vitro strong> and in vivo activities of IgG and F(ab′)2 fragments of a mixture of three monoclonal anti-her-2 antibodies. J. Exp. Clin. Cancer Res. 2004;10(10):3542–51. doi:10.1158/1078-0432.CCR-03-0549.
- Sreedhara A, Lau K, Li C, Hosken B, Macchi F, Zhan D, Shen A, Steinmann D, Schöneich C, Lentz Y. Role of surface exposed tryptophan as substrate generators for the antibody catalyzed water oxidation pathway. Mol Pharm. 2013;10:278–88.
- Subelzu N, Schöneich C. Pharmaceutical excipients enhance iron-dependent photo-degradation in pharmaceutical buffers by near UV and visible light: tyrosine modification by reactions of the antioxidant methionine in citrate buffer. Pharm Res. 2021;38(5):915–30. doi:10.1007/s11095-021-03042-8.
- Garcia NK, Sreedhara A, Deperalta G, Wecksler AT. Optimizing hydroxyl radical footprinting analysis of biotherapeutics using internal standard dosimetry. J Am Soc Mass Spectrom. 2020;31(7):1563–71. doi:10.1021/jasms.0c00146.
- Uehara H, Rao VA. Metal-mediated protein oxidation: applications of a modified ELISA-based carbonyl detection assay for complex proteins. Pharm Res. 2015;32:691–701.