
ABSTRACT
In the European Union, the use of veterinary drugs belonging to the A6 group is prohibited in food-producing animals according to Commission Regulation (EU) No. 2010/37. The aim of this study was to improve the analytical control strategy by developing a single method to analyse residues of prohibited pharmacologically active substances in milk. For this, a single method was developed to analyse 16 prohibited pharmacologically active substances belonging to five different substance classes at required or recommended levels: nitroimidazoles at 3 μg kg−1, nitrofurans at 0.5 μg kg−1, chloramphenicol at 0.1 μg kg−1, dapsone at 5 μg kg−1 and chlorpromazine at 1 μg kg−1. Milk sample preparation started with an acid hydrolysis combined with a derivatisation. These steps were followed by a clean-up consisting of a dispersive solid-phase extraction and a liquid–liquid extraction. Finally, the sample extracts were analysed by liquid chromatography combined with tandem mass spectrometry, operating alternately in the positive and negative mode. The method was fully validated according to Commission Decision 2002/657/EC for bovine milk and additionally validated for caprine milk. The validation proved that the method is highly effective to detect and confirm all 16 substances in bovine and caprine milk and, additionally to quantify 15 of these substances in bovine milk and 13 of these substances in caprine milk. This study resulted in a new multi-class method to detect, quantify and confirm the identity of 16 prohibited pharmacologically active substances belonging to five different substance classes in two types of milk.
Introduction
The European Union established regulations to control and enforce veterinary drug use in food-producing animals (e.g. milk-producing bovine and caprine) (European Commission Citation2017) because of potential public health risks (Baynes et al. Citation2016). These risks include direct exposure to toxic veterinary drug residues and related metabolites and the chance of developing antimicrobial resistance in bacteria. Strict legislation regarding the application of veterinary drugs and their enforcement are established in the European Union: Regulation (EU) 2017/625 (European Commission Citation2017) and Commission Regulation (EU) No. 2010/37 (European Commission Citation2010). Commission Regulation (EU) No. 2010/37 (European Commission Citation2010) includes, in addition to maximum residue limits for registered veterinary drugs, a table with prohibited veterinary drugs, classified as A6 group in Council Directive 96/23/EC Annex 1 (European Commission Citation1996). In this paper these substances are referred to as prohibited pharmacologically active substances (PPAS). The PPAS group evaluated in this study includes nitroimidazoles, nitrofurans, chloramphenicol, dapsone and chlorpromazine.
Nitroimidazoles (NIZs) are prohibited because concerns have been raised about toxicity, mutagenicity and genotoxicity of this substance class (WHO Citation1987; Boechat et al. Citation2015). However, NIZs are important antiparasitic agents with a high biological activity (Boechat et al. Citation2015). The NIZs mentioned in Commission Regulation (EU) No. 2010/37 (European Commission Citation2010) are metronidazole (MNZ), ronidazole (RNZ) and dimetronidazole (DMZ). In addition, the corresponding metabolites hydroxy-metronidazole (MNZ-OH) and 2-hydroxymethyl-1-methyl-5-nitroimidazole (HMMNI) are included in this study. HMMNI is the hydroxyl metabolite of both RNZ and DMZ. Finally, ipronidazole (IPZ), hydroxy-ipronidazole (IPZ-OH) and ternidazole (TNZ) are also included in this study.
The second veterinary drug class of interest is the nitrofurans (NFs), which includes furazolidone, furaltadone, nitrofurantoin, nitrofurazone and nifursol. These substances are broad spectrum antimicrobial agents, with genotoxic and carcinogenic properties (EFSA Citation2015) and therefore prohibited. The NFs rapidly metabolise to 3-amino-2-oxazolidinone (AOZ), 3-amino-5-methylmorpholino-2-oxazolidinone (AMOZ), 1-aminohydantoin (AHD), semicarbazide (SEM) and 3,5-dinitrosalicylic acid hydrazide (DNSH), respectively (Molognoni et al. Citation2021). For that reason, instead of the native substances, the metabolites are included in the analyses of NFs in food products as mentioned in the guideline created by the European advisory body, the European Reference Laboratory (EURL) (CRL Citation2007).
Three other relevant PPAS are chloramphenicol (CAP), dapsone (DAP) and chlorpromazine (CPZ). CAP is a broad-spectrum antibiotic and has been prohibited as results of concerns about its genotoxicity, embryotoxicity and fetotoxicity, carcinogenicity and the possible contribution to aplastic anaemia (JECFA Citation2004; Baynes et al. Citation2016). DAP is under debate because of its carcinogenic and genotoxic properties (EFSA Citation2005; EMA Citation2012). The lack of information about reproductive toxicity and teratogenicity has made DAP a PPAS since 1996 (EMA Citation1996b). CPZ belongs to the class of tranquillizers. Prohibition of CPZ is based on the advice of the Joint FAO/WHO Expert Committee on Food Additives (JECFA) because of, among other things, the lack of relevant toxicological data (JECFA Citation1991; EMA Citation1996a).
The EURLs responsible for the analysis of PPAS have created guidelines for the analysis of most of these substances in food, including a recommended detection level in all kinds of food products, including milk (CRL Citation2007). The recommended detection level for the three prohibited NIZs including the corresponding metabolites is 3 µg kg−1 and for DAP 5 µg kg−1. For NFs and CAP the guidelines refers to the regulation where a legal level is established, namely the minimum required performances limit (MRPL). For NFs and CAP the legislation, Commission Decision 2002/657/EC (European Commission Citation2002) appointed a MRPL. The MRPL has to be considered as the reference point of action (RPA) according to Commission Decision 2005/34/EC (European Commission Citation2005). Recently, the RPA levels were revised; the RPA for NFs will be 0.5 µg kg−1 and for CAP 0.15 µg kg−1 valid from 28 November 2022 as described in Commission Regulation (EU) 2019/1871 (European Commission Citation2019). For CPZ, no recommended detection level or RPA is established yet.
The analysis of PPAS requires highly sensitive, selective and accurate methods. Many papers have been published on the analysis of a single class of PPAS residues (or in combination with regulated substances) in milk (e.g. NIZs (Thompson et al. Citation2009; Gui et al. Citation2011; Tölgyesi et al. Citation2012; Hernández-Mesa et al. Citation2014, Citation2015, Citation2017, Citation2018; Mitrowska et al. Citation2014; Tuzimski and Rejczak Citation2017; Wang et al. Citation2019), NFs (Ryan et al. Citation1975; Galeano Dı́az et al. Citation1997; Chu and Lopez Citation2007; Rodziewicz Citation2008; Alkan et al. Citation2016; Śniegocki et al. Citation2018), CAP (Allen Citation1985; Sørensen et al. Citation2003; Ferguson et al. Citation2005; Nicolich et al. Citation2006; Mohamed et al. Citation2007; Rezende et al. Citation2012; Berlina et al. Citation2013), DAP (Suhren and Heeschen Citation1993; Van Rhijn et al. Citation2002; Hadjigeorgiou et al. Citation2009; Kaklamanos and Theodoridis Citation2012; Varenina et al. Citation2016) and CPZ (Ohkubo et al. Citation1993)), of which most use a chromatographic technique coupled with mass spectrometric detection. In order to analyse all desired PPAS, five different single class methods are needed. In the last twenty years, analysis methods have been published to analyse all kinds of combinations of PPAS residues (in combination with regulated substances) in milk e.g. NIZs combined with DAP (Ortelli et al. Citation2009), NIZs with CAP (Cronly et al. Citation2010; Wang et al. Citation2016), NIZs with CPZ and CAP (Zhan et al. Citation2012) and NIZs with CPZ, CAP and DAP (Kibechu and Sichilongo Citation2012; Robert et al. Citation2013; Amelin et al. Citation2018; Jadhav et al. Citation2019). However, multiple methods for the analyses of NFs together with other substance classes are limited not only in milk but also in all kind of matrices (Perez et al. Citation2002; Xia et al. Citation2008; Shen et al. Citation2013; Kaufmann et al. Citation2015; Shendy et al. Citation2016; Zhang et al. Citation2017; Aldeek et al. Citation2018; Chen et al. Citation2020). For NFs analysis, a hydrolysis is required to include protein-bound residues and a derivatisation is applied to stabilise the metabolites and to enhance the mass spectrometric signal (Molognoni et al. Citation2021). However, using these hydrolysis and derivatisation only one validated multi-method for milk analysis is published (Kaufmann et al. Citation2015) and this method is a combination of only the NF metabolites and CAP.
To our knowledge, no fully validated method has previously been reported, including PPAS belonging to five different substance classes in milk using ultra high-performance liquid chromatography-tandem mass spectrometry (LC-MS/MS). This study describes a new multi-class method and its validation for the detection, quantification and confirmation of identity of the PPAS in milk. This new approach yields a far more cost-effective surveillance of the PPAS in milk using one multi-class method instead of multiple single-class methods.
Material and methods
Reagents and equipment
Milli-Q water was prepared using a Milli-Q system at a resistance of at least 18.2 MΩ cm (Millipore, Billerica, MA, USA). Ultra LC-MS grade methanol, acetonitrile and ethyl acetate were obtained from Actu-All Chemicals (Oss, The Netherlands). Hydrochloric acid (37%), trisodium phosphate dodecahydrate, sodium hydroxide, acetic acid (100%) and formic acid (100%) were obtained from Merck (Darmstadt, Germany). 2-Nitrobenzaldehyde and ammonium formate were obtained from Sigma Aldrich (St. Louis, MO, USA) and AOAC dispersive solid-phase extraction (SPE) 15 mL kit (400 mg primary-secondary amine (PSA), 400 mg C18 and 1200 mg MgSO4) was obtained from Agilent technologies (Santa Clara, CA, USA).
Hydrochloric acid solution (0.2 mol L−1) was prepared by diluting 17 mL hydrochloric acid (37%) in 1 L Milli-Q. 2-Nitrobenzaldehyde solution (100 mmol L−1) was prepared by dissolving 1.51 g 2-nitrobenzaldehyde in 100 mL methanol. Trisodium phosphate solution (0.3 mol L−1) was prepared by dissolving 11.4 g trisodium phosphate dodecahydrate in 100 mL Milli-Q. Sodium hydroxide solution (2 mol L−1) was prepared by dissolving 8.0 g sodium hydroxide in 100 mL Milli-Q. The reconstitution solution was prepared by mixing 20 mL acetonitrile with 80 mL Milli-Q and 100 µL acetic acid. Ammonium formate solution (1 mol L−1) was prepared by dissolving 6.3 g ammonium formate (1 mol L−1) in 100 mL Milli-Q. Mobile phases A and B were prepared by diluting 2 mL ammonium formate solutions and 160 µL formic acid to 1 L with Milli-Q and methanol, respectively.
The reference standards of MNZ, RNZ, DMZ, MNZ-OH, CAP, DAP, and CPZ were obtained from Sigma Aldrich. HMMNI, IPZ, IPZ-OH, TNZ HCl, AMOZ, AHD HCl and SEM HCl were obtained from Witega (Berlin, Germany). AOZ was obtained from Santa Cruz Biotechnology (Dallas, TX, USA). DNSH was obtained from Toronto Research Chemicals (Toronto, ON, Canada).
The isotopic labelled standards MNZ-d4, DAP-d8, and DNSH-15N2 were obtained from Toronto Research Chemicals (Toronto, ON, Canada). RNZ-d3 and AHD-13C3 were obtained from Sigma–Aldrich (St. Louis, MO, USA). DMZ-d3, HMMNI-d3, IPZ-d3, IPZ-OH-d3, AOZ-d4, AMOZ-d5, SEM-13C15N2, CAP-d5, and CPZ-d6 HCl were obtained from Witega (Berlin, Germany).
Stock solutions were prepared at 100 mg L−1 in methanol for most standards and internal standards. However, DAP and DAP-d8 were prepared at 1000 mg L−1 in methanol and CPZ and CPZ-d6 were prepared at 100 mg L−1 and 1000 mg L−1, respectively, in ethanol. From the stock solutions, a mix solution was prepared at 3 mg L−1 NIZs, 0.5 mg L−1 NFs (DNSH was added separately), 0.1 mg L−1 CAP, 5 mg L−1 DAP, and 1 mg L−1 CPZ in Milli-Q. This mix solution and an individual solution of 0.5 mg L−1 DNSH were diluted 20 times in Milli-Q water to achieve the final standard solution (5–250 µg L−1). In addition, a mix solution of internal standards was prepared at 3 mg L−1 NIZs, 1 mg L−1 NFs, 0.3 mg L−1 CAP-d5, 5 mg L−1 DAP-d8, and 1 mg L−1 CPZ-d6, followed by a twenty-time dilution in Milli-Q water to a final concentration of 15–250 µg L−1.
Optimised sample preparation
The final optimised sample preparation procedure is based on the method published by Mulder et al. (Citation2005). The procedure for the sample preparation is as follows: 2.0 ± 0.05 g of homogenised raw milk was weighed into a 12 mL polypropylene tube and 80 µL of internal standard solution (15–250 µg L−1) was added. Subsequently, 5 mL hydrochloric acid solution and 50 µL 2-nitrobenzaldehyde solution were added. The sample solution was shaken head-over-head (Heidolph REAX-2, Schwabach, Germany), overnight at 37°C to hydrolyse protein-bound NF metabolites and to derivatise the metabolites into their nitrophenyl (NP)-derivatives. After cooling of the sample solution to room temperature, 500 µL of trisodium phosphate solution and at least 300 µL of sodium hydroxide solution were added to adjust the pH to 7.0 ± 0.5. Next, the sample solution was diluted with 10 mL Milli-Q water and transferred into a 50 mL polypropylene tube containing the AOAC dispersive SPE kit. The tube was shaken head-over-head for 5 minutes and afterwards the sample solution was centrifuged for 10 minutes at 3000 g (MSE Falcon 6/300, Heathfield, UK). After centrifugation, the sample solution was transferred into a clean 50 mL polypropylene tube. Additionally, a liquid–liquid extraction was performed by adding 8 mL of ethyl acetate to the sample solution and subsequently this was shaken head-over-head for 20 minutes. After centrifugation (3000 g, 10 min), the ethyl acetate upper layer was transferred into a 12 mL glass tube. The liquid–liquid extraction procedure was repeated, and the ethyl acetate layers were combined. The ethyl acetate was evaporated under nitrogen at 40°C (TurboVap LV Evaporator Zymark, Hopkinton, MA, USA), and the remaining part was dissolved in 200 μL reconstitution solvent (0.1 (v/v)% formic acid in 20 (v/v)% acetonitrile). Finally, the sample extraction was centrifuged (3000 g, 10 min) and transferred into a vial to facilitate analysis by LC-MS/MS.
Method development
During the method development, several sample preparation steps were studied and optimised. The individual sample preparation steps were first evaluated aiming for high absolute recoveries. The absolute recovery is the relative response of a sample spiked before sample preparation compared to a sample spiked after sample preparation. The absolute recovery could not be established for the NFs because no derivatised NF marker metabolite standards were available for spiking the samples after sample preparation. Additionally, the optimisation was evaluated based on the signal-to-noise ratio of the observed peaks in the chromatogram, for all substances including the NFs. The signal-to-noise ratio was automatically determined by the software during processing of the data.
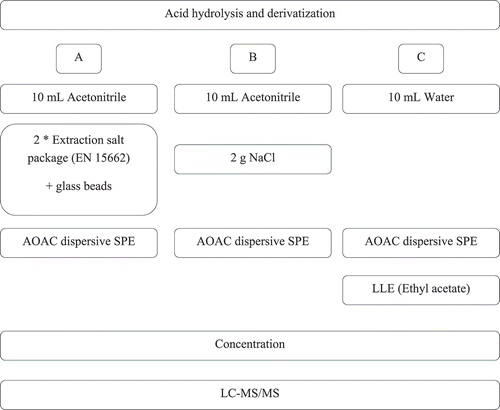
In the first experiment, three different clean up procedures were selected () based on in-house experience and expectations from the literature. Procedure A is in principle based on a method for prohibited veterinary drugs in urine, as published by León et al. (León et al. Citation2012). In the procedure, the enzymatic hydrolysis as applied by León was omitted since insignificant glucuronidation of CAP was expected in milk (Nouws et al. Citation1986). On the other hand, an acidic hydrolysis and derivatisation step were added for the NFs. These steps were followed by the capture of the water content by MgSO4, included in the extraction salt package (EN 15662). In procedure B, the same procedure was applied, but the water content was not captured by MgSO4 but separated from the acetonitrile by using 2 g NaCl. In both procedures A and B, the acetonitrile layer was cleaned with an AOAC dispersive SPE kit and concentrated. Procedure C is described in the section optimised sample preparation above.
Figure 1. A schematic representation of the different sample clean up procedures tested during method development. LLE is liquid-liquid extraction using ethyl acetate
A second experiment was performed to optimise procedure C regarding the individual constituents of the AOAC dispersive SPE kit. In this experiment, the sample preparation was performed as described in the optimised sample preparation section, only the use of the AOAC dispersive SPE kit was adjusted. The SPE kit was replaced by the individual constituents (PSA, C18, and MgSO4), all possible combinations of these constituents, or was completely excluded. The absolute recoveries and signal-to-noise ratios were used to determine the influence of all the individual constituents and their interactions. Statistical evaluation of the absolute recoveries was carried out using a factorial design as described by Andries and de Vries (Andries and De Vries Citation2007).
In experiment three, the sample preparation was performed as described in the optimised sample preparation section, and the ethyl acetate liquid–liquid extraction was optimisation. Four different variations were tested. After the described dispersive SPE clean-up, the substances were extracted using a single or double liquid–liquid extraction procedure with 8 or 6 mL ethyl acetate.
LC–MS/MS analysis
After sample preparation, the LC-MS/MS analysis was carried out on an Acquity UPLC (Waters, Milford, MA, USA) or a Nexera UHPLC (Shimadzu, Kyoto, Japan). An Acquity UPLC BEH C18 analytical column (Waters, Milford, MA, USA) of 100 × 2.1 mm with a particle size of 1.7 µm, was placed in a column oven at 35°C. Into the LC column, 10 µL of the sample extract was injected. The substances were chromatographically separated by gradient elution using a flow rate of 0.4 mL min−1. The gradient started at 100% mobile phase A and linearly increased to 30% mobile phase B in 4 minutes. The next 2 minutes were isocratic, followed by an increase of the percentage mobile phase B to 70% in 1.5 min and to 100% in the next 1.5 min, with a final hold of 1.0 min and a re-equilibration time of 2 min at 100% A.
After the chromatographic separation, the substances were introduced directly into a Q-Trap 6500+, Q-Trap 6500 or Q-Trap 5500 mass spectrometer (Sciex, Framingham, MA, USA). The mass spectrometer was operated in polarity switching mode (i.e., alternately in positive and negative electrospray ionisation mode). The operating parameters were curtain gas flow 40 psi (N2), nebulising gas flow 50 psi (N2), heater gas flow 50 psi (N2), source temperature 400°C and ion spray voltage (-) 4500 V. The precursor ions were fragmented to product ions using collision induced dissociation (N2). The scheduled Multiple Reaction Monitoring (MRM) transitions (60 s window in positive and 120 s window in negative mode) are presented in . Data were processed using the Multiquant software V2.1.1 (Sciex, Framingham, MA, USA), resulting in a response of each transition. The response was corrected using the corresponding isotopically labelled internal standards. Since no labelled internal standards were available for MNZ-OH and TNZ, HMMNI-d3 was used for these two substances. The response factors were calculated by dividing the area of the most abundant product ion of the substance by the area of the internal standard.
Table 1. MRM transitions of the PPAS and their internal standards, including the declustering potential (DP), collision energy (CE) and collision cell exit potential (CXP). The underlined product ion is the most abundant one
Method validation
The method was fully validated for bovine milk, according to the criteria described in Commission Decision 2002/657/EC (European Commission Citation2002). The validation was carried out at three days by two different technicians, on three different LC-MS/MS systems and included bovine milk samples (n = 24) sampled from milk tanks. The milk samples were collected by the Dutch Food and Consumer Product Safety Authority on different, randomly selected farms at various time points during the year. Ideally, a validation is carried out using incurred certified reference materials. However, these materials are not available for this specific application; therefore, blank samples were spiked as an alternative.
The spike level or the validation level of NIZs was 3 µg kg−1 and DAP was validated at 5 µg kg−1 as recommended for milk by the EURLs (CRL Citation2007). Recently, the RPA for NFs and CAPs was revised by Commission Regulation (EU) 2019/1871 (European Commission Citation2019), namely 0.5 µg kg−1 for NFs and 0.15 µg kg−1 for CAP. Since the validation occurred before the establishment of these new RPAs, the validation was carried out at 0.5 µg kg−1 for NFs and 0.1 µg kg−1 for CAP. As a result, for CAP, the highest spike level corresponds to the revised RPA (European Commission Citation2019). Finally, 1 µg kg−1 was used as the validation level of CPZ because no RPA or recommended concentration exists.
During the validation, blank samples were spiked at three different levels: 0.5, 1.0 and 1.5 times validation level. Note that Commission Decision 2002/657/EC (European Commission Citation2002) states to use 1.0, 1.5 and 2.0 times the MRPL for prohibited substances, but because lower levels are more relevant for enforcement, validation levels were lowered. In addition, a detection capability as low as possible is preferred for PPAS because of their zero-tolerance policy.
The following validation parameters are related to a quantitative confirmatory method and were determined: selectivity, linearity, trueness, repeatability (RSDr), repeatability including matrix variation (RSDr*), within-laboratory reproducibility (RSDRL), decision limit (CCα), detection capability (CCβ), confirmation of the identity, stability and robustness.
Selectivity
The selectivity was determined using 21 blank bovine milk samples analysed with the addition of only the internal standards. Selectivity was assessed by evaluating the signal of the blank materials for interferences at the retention times corresponding to the PPAS.
Linearity
On three different validation days, a matrix matched calibration line was prepared by adding standard solution of the PPAS (5–250 µg L−1) to aliquots of a blank bovine milk sample at 0, 0.25, 0.5, 1.0, 1.5 and 2.0 times the validation level. The added concentration of a substance was plotted against the internal standard corrected response and the response factor (as explained in section LC–MS/MS analysis). Least squares linear regression was used to create a matrix matched calibration line. The linearity of this line was accepted if the coefficient of correlation was at least 0.990.
Trueness, repeatability and within-laboratory reproducibility
Trueness and repeatability, including matrix vari-ation (RSDr*) and within-laboratory reproducibility (RSDRL), were determined using seven different blank bovine milk samples (other samples than the three used for the matrix matched calibration lines) on each individual day. These samples were spiked at 0.5, 1.0 and 1.5 times the validation level, prepared and analysed. In addition, the true repeatability (RSDr) was determined as described in Commission Decision 2002/657/EC (European Commission Citation2002), using seven aliquots of a single blank bovine milk sample. These aliquots were spiked at validation level, prepared and analysed. The whole procedure was repeated on two more days, to obtain 21 results for each validation level with different milk samples and 21 results of the same milk sample. Concentrations were calculated using the matrix matched calibration line prepared and analysed under the same circumstances. The average measured concentration of the spiked samples was divided by the theoretical added concentration, resulting in the trueness. The RSDr*, RSDRL, and RSDr were calculated using analysis of variance (ANOVA).
According to Commission Decision 2002/657/EC (European Commission Citation2002), the trueness of the method should comply with the established criteria for a quantitative analysis. The criteria depend on the validation levels: for levels between 1 and 10 µg kg−1, a trueness of 70%–110% is accepted and for levels lower than 1 µg kg−1 a trueness of 50%–120% is accepted (European Commission Citation2002). The relative within-lab reproducibility (RSDRL) is accepted below the value calculated from the Horwitz equation (Horwitz et al. Citation1980). However, the Horwitz equation is not applicable to the lower concentration range (<120 μg kg−1) (Thompson Citation2000) and therefore a complementary model was suggested. We adopted these more stringent criteria of Thompson. Based on these criteria, the RSDRL and RSDr are accepted below 22% and 14.7%, respectively. For RSDr* the same criteria were applied as for RSDr in this validation, being a worst-case approach.
Decision limit (CCα) and detection capability (CCβ)
For a quantitative confirmatory method, the decision limit (CCα) and detection capability (CCβ) have to be established (European Commission Citation2002). Because zero tolerance applies for all PPAS, the decision limit CCα (α-error is 1%) indicates the lowest level at which substances can be detected and confirmed (European Commission Citation2002). Results above the CCα should be considered to be non-compliant. However, for NFs and CAP, a second CCα (α-error is 5%) has to be determined because these substances have a legally established reference point of action (RPA) (European Commission Citation2019). In this case, the CCαRPA based on the RPA means the limit at and above which it can be concluded with an error probability of α that the actual quantitative result of the sample is above the RPA. However, results below RPA but above CCα based on zero tolerance should be reported to the competent authority as the authority has to retain a record of these findings in case of recurrence.
The CCα and CCβ based on zero tolerance were calculated with the calibration curve procedure according to ISO 11843 (European Commission Citation2002). According to the EURL guidelines, CCα should be lower than the recommended detection concentration or the minimum required performances limit, and thus, the RPA for a confirmatory method (CRL Citation2007). In this validation, the method is considered as applicable for the detection of PPAS if both CCα and CCβ are below the MRPL, RPA or concentration recommended by the EURL.
The CCαRPA and CCβRPA were calculated according to the procedure described in Commission Decision 2002/657/EC (European Commission Citation2002) specified for registered veterinary drugs with a maximum residue limit. The procedure includes the reproducibility of spiked samples at the RPA.
If one of the quantitative validation parameters does not comply for a certain substance, the method is not applicable for quantitative analysis of this specific substance. However, for such a substance a qualitative approach can be used. The detection capability can be determined by the investigation of fortified blank material at and above the decision limit. In this case, the concentration level, where only ≤ 5% (n is minimal 20) false compliant results remain, equals the detection capability of the method (European Commission Citation2002). In other words, at least 20 of the 21 samples with addition at the relevant detection level showed a response with a minimum signal-to-noise ratio of 10.
Confirmation of identity
Another validation parameter relates to the confirmation of the substance identity. The substance identity is confirmed if the relative retention time of that substance deviates at maximum 2.5% compared to the average relative retention time of the same substance in the matrix matched calibration standards. Furthermore, the relative abundance of both product ions (ion ratio) should not deviate from the matrix matched calibration standards more than the criteria described in Commission Decision 2002/657/EC (European Commission Citation2002). Over 95% of the validation samples should comply with these confirmatory criteria, as required for a confirmatory analysis.
Stability
For quantitative analysis, it is important to study the stability of the substances included. In the stock solution, the stability was determined and evaluated following the procedure and criteria as proposed by Berendsen et al (Berendsen et al. Citation2011). In addition, the stability in the final extract was established by re-analysing seven extracts (of one day only at 1 times validation level) after one and a half week of storage in the freezer. For acceptance, the stability of the extracts, the trueness and RSDr* still need to comply with the validation criteria as mentioned before to confirm that no significant degradation has occurred.
Robustness
The robustness of the method has become apparent from the method development, whereby minor and major variations have been made and the influences are discussed in the method development section.
Additional validation
An additional validation was performed for caprine milk at validation level, using seven different caprine milk samples. Caprine milk was quantified based on a matrix matched calibration line made from bovine milk because it would be far more efficient in practice if bovine and caprine milk can be run in a single series with a single set of quality control samples. The validation parameters selectivity, trueness, repeatability including matrix variation (RSDr*) and confirmation of the identity were determined and assessed as described for bovine milk. The within-laboratory reproducibility (RSDRL) was estimated based on the RSDr*, using the equation RSDRL = 1.6 times RSDr* (van Reeuwijk Citation1998). Finally, if all validation parameters comply, the CCα is considered to be below the validation level and CCβ will be equal to the validation level because the additional validation included insufficient data points for a calculation of CCα and CCβ.
Results and discussion
Method development
Three different experiments were performed to develop a method for the analysis of PPAS in milk. The first experiment studied three different clean up procedures; A, B and C (). In procedure A, we encountered practical issues during the removal of the high water content after derivatisation. The water content was removed by MgSO4 present in two extraction salt packages. However, this resulted in agglomeration of the MgSO4, even with the use of glass beads, which complicated mixing and homogenising of the sample solution. Also, in procedure B practical issues were encountered. In this procedure, the water was not removed but separated from the acetonitrile by using NaCl. In the acetonitrile layer unwanted gelation occurred. The gel formation made the acetonitrile transfer impractical and reduced the effective volume that could be transferred and used in the next steps. No practical issues were encountered during the sample preparation of procedure C, making it the preferred procedure (from the practical perspective) even though this procedure consists of more steps, including a time-consuming ethyl acetate liquid–liquid extraction. Time can be saved for a large sample series by rapidly freezing (approx. 20 minutes) the aqueous part at < −70 °C and immediately decanting the ethyl acetate layer.
The absolute recoveries of the three procedures for all substances except NFs were compared (). The NF recoveries could not be established because no derivatised NF metabolite standards were available. The evaluation of the NFs was performed based on the signal-to-noise ratios, but as the signal-to-noise ratio is relevant to obtain detection limits as low as possibly achievable, signal-to-noise ratios for all substances were evaluated (Supplementary information (SI) Table S1). For all substances, absolute recoveries (NFs are not included) of procedure B were lower than for procedures A and C (). Absolute recoveries of procedures A and C were in the same order of magnitude. Note that DMZ, MNZ-OH, HMMNI, DNSH and CAP showed in generally relatively low signal-to-noise ratios and, therefore, a sufficient recovery for especially these substances is crucial. Even though absolute recoveries for some substances were below 65%, this was sufficient to detect them at the relevant levels. The results of the absolute recovery of procedure A are comparable with the results published by Kibechu et al. who use a similar AOAC clean-up (only without hydrolysis and derivatisation) in milk. The absolute recoveries in that research vary between 33% and 71% for MNZ, RNZ, DMZ, CAP, DAP and CPZ (Kibechu and Sichilongo Citation2012). In our method, for CPZ only the absolute recovery was severely higher in procedure A compared to procedure C. Nevertheless, for all substances procedure C showed higher or comparable signal-to-noise ratios compared to procedure A, also for the NFs (SI Table S1). Based on this observation and the limited practicability of procedure A, procedure C was preferred. Using procedure C detection of all PPAS at low levels was achievable. Therefore, the focus was on further optimisation of procedure C.
Figure 2. Absolute recoveries of procedure A (white), B (light grey) and C (black) for PPAS at 1 µg kg−1; chloramphenicol at 0.3 µg kg−1. The nitrofurans are not included because no derivatised marker metabolite standards were available
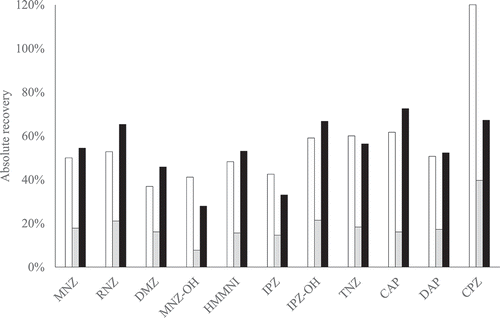
In procedure C, an AOAC SPE kit was included in the sample preparation. Using the AOAC SPE kit is not straightforward in an aqueous environment because some substances included in this method might have affinity to the C18 material present in the kit. Nevertheless, the first experiment surprisingly showed that the absolute recoveries and signal-to-noise ratios of the substances were sufficient for this method to detect all substances at relevant levels. To even further improve the method, the influence of PSA, C18 and MgSO4 in the kit was studied. In most combinations of the individual kit constituents, an unwanted gel was formed during the ethyl acetate liquid–liquid extraction. Similarly, a thick gel was formed during the sample preparation without the addition of PSA, C18 or MgSO4. This set-up (a hydrolysis and derivatisation followed by liquid–liquid extraction with ethyl acetate, without the addition of PSA, C18 or MgSO4) is similar to the method published by Kaufmann et al. (Citation2015). Kaufman also observed an emulsion in the ethyl acetate layer using milk as matrix, but in the published method the liquid–liquid extraction was followed by a further clean-up using SPE, even though the final extracts could still be very cloudy. On the other hand, in the method published by Rodziewicz (Citation2008) sample preparation of milk was also performed with a hydrolysis and derivatisation followed by liquid–liquid extraction with ethyl acetate. The author did not mention any practical issues, this might be due to the fact that the raw milk was centrifuged and the upper fat layer was removed beforehand (Rodziewicz Citation2008). Fat removal of raw milk was also performed by Alkan et al. (Citation2016) prior to hydrolysis, derivatisation and liquid–liquid extraction and again no practical issues were mentioned.
In our second experiment only four combinations of constituents resulted in a practical sample preparation: the AOAC SPE kit, self-mixed PSA, C18 and MgSO4, the combination of C18 and MgSO4, and only C18. Apparently, the gel formation is reduced by the presence of C18, except for the combination of PSA and C18. No explanation has been found for this observation.
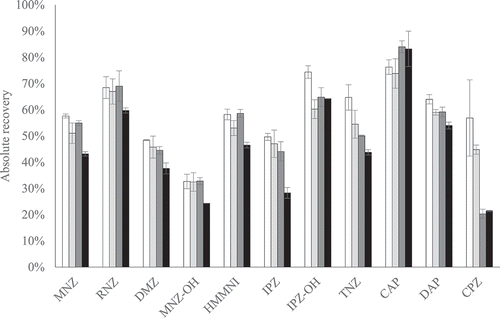
The absolute recoveries of the four combinations without practical implications are presented in . In general, the absolute recoveries are lower in the sample preparation which included only C18, as could be expected because most substances have affinity with C18 in aqueous environment. In particular, it can be expected that the absolute recovery of CPZ is negatively influenced since this substance is the most lipophilic substance included. Indeed, lower absolute recoveries of CPZ were observed for the sample preparation with only C18 (21 ± 0.2%) and also with C18 and MgSO4 (20 ± 2%) compared to absolute recoveries of the sample preparation with the AOAC SPE kit (57 ± 15%) or self-mixed PSA, C18 and MgSO4 (45 ± 2%) (). For CPZ, the negative effect on the absolute recovery of C18 material was statistically confirmed (p-value = 0.00002). However, the combination of all three constituents resulted in higher absolute recoveries compared to using only C18 or C18 and MgSO4. Apparently, PSA influences the affinity of CPZ to C18 material as becomes apparent from the significant interaction of C18 and PSA (p-value = 0.0018). Thus, the combination of PSA, C18 and MgSO4 is needed to prevent gel formation and it results in the overall highest absolute recoveries. In addition, the signal-to-noise ratios as shown in SI Table S2 hardly differ regardless of the constituents used in the preparation. Therefore, the sample preparation with the AOAC SPE kit is preferred or the cheaper alternative self-mixed PSA, C18 and MgSO4.
Figure 3. The average absolute recovery of sample preparation with the AOAC SPE kit (white) or this kit replaced by 400 mg PSA, 400 mg C18 and 1200 mg MgSO4 (light grey), by 400 mg C18 and 1200 mg MgSO4 (dark grey) or by 400 mg C18 (black), for all PPAS at validation level and half the validation level, except the nitrofurans. The error bars indicate ± standard deviation (n = 2)
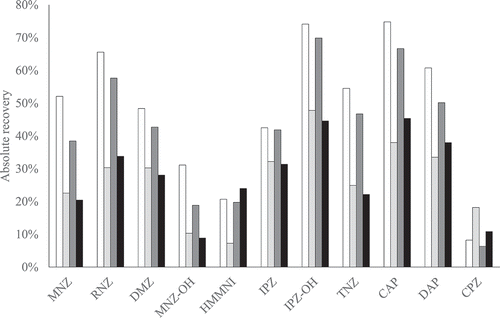
In experiment three, the liquid–liquid extraction was optimised. For both the volume and the number of replications, the absolute recovery was determined, as well as the signal-to-noise ratio ( and SI Table S3). The absolute recovery is in general higher when using a second extraction and a little bit higher using two times 8 mL instead of two times 6 mL ethyl acetate. Remarkable, CPZ shows an unexpected result. For this substance, the single extraction shows higher absolute recoveries and signal-to-noise ratios compared to the duplicated extraction. No explanation has been found for this observation. However, the negative influence of the double extraction on the CPZ response does not outweigh the positive effect for all other PPAS. Therefore, the final optimised method consists of a double liquid–liquid extraction using 8 mL ethyl acetate.
Figure 4. Absolute recoveries of sample preparation with a duplicated (white) or single (light grey) ethyl acetate extraction with 8 mL or a duplicated (dark grey) or single (black) ethyl acetate extraction with 6 mL for PPAS at 1 µg kg−1; CAP at 0.3 µg kg−1. The NFs are not included because no derivatised marker metabolite standards were available
Validation
The aim of the validation was to assess the quantitative confirmatory aspect of the method. During the validation, blank bovine milk samples were spiked at 0.5, 1.0 and 1.5 times validation level. An example of chromatograms from bovine milk spiked at 1.0 times validation level is presented in SI for all PPAS. presents an overview of the validation results for bovine milk. The validation parameters were calculated on the basis of 21 results (seven samples per validation level on three different days). Only for IPZ, AOZ, AHD and SEM, 20 results were used at 0.5 times validation level because a single sample showed severe chromatographic fluctuations. These fluctuations resulted in substances eluting outside of the detection window, so no data was obtained. For the following analysis, the scheduled MRM windows were broadened to overcome this problem.
Table 2. The validation results in bovine milk; trueness, repeatability (RSDr), repeatability including matrix variance (RSDr*), within-laboratory reproducibility (RSDRL), decision limit (CCα), detection capability (CCβ) based on zero tolerance and CCαRPA and CCβRPA based on the RPA. n is the number of samples used for the calculations and is only presented if lower than 21. The underlined values exceed the established criteria
Selectivity
Selectivity was assessed by investigating the signal of the blank materials for interferences. No interfering signals were observed in the blank samples at the retention times corresponding to the transitions of the substances to be validated. Therefore, the selectivity of the method is considered sufficient.
Linearity
The matrix match calibration line was injected at the beginning and at the end of the sample series. The linearity of the line was expressed by the coefficient of correlation. In all cases, the coefficient of correlation complied with the criterion: they were between 0.992 and 1.000. For this reason, the linearity of all substances is accepted in the range between 0.25 and 2 times the validation level.
Trueness, repeatability and within-laboratory reproducibility
shows the results of the validation parameters trueness, true repeatability (RSDr), repeatability including matrix variation (RSDr*) and within-laboratory reproducibility (RSDRL). Following the validation criteria in Commission Decision 2002/657/EC (European Commission Citation2002), a trueness of 70%–110% is accepted for levels between 1 and 10 µg kg−1 a trueness of 50%–120% for levels lower than 1 µg kg−1. For all substances except for CPZ the trueness was between 95 and 107%, and thus the trueness complies with the criteria for all substances other than CPZ. The trueness of CPZ was above 120% for two validation levels (1 µg kg−1 and 1.5 µg kg−1). This criterion is probably exceeded because of the sub-optimal sensitivity of one LC-MS/MS instrument for CPZ. Therefore, the validation did not prove the applicability of the method to quantify CPZ. However, CPZ can be analysed qualitatively using the reported method. Overall, it can be stated that the qualitative screening of PPAS is more important than the quantification because the use of PPAS is prohibited overall. In case of a suspect sample for CPZ, a quantitative result can be obtained using a validated single substance method or by applying multiple level standard addition.
The RSDr was between 2.2% and 13.5% for all substances and at all validation levels, thus below the limit of 14.7%. Therefore, the RSDr is accepted. Also, the RSDr* and RSDRL, comply with the established criteria for all substances and at all levels. (Kaufmann et al. Citation2015)
Decision limit (CCα) and detection capability (CCβ)
For all PPAS that complied with the quantitative performance criteria, CCα and CCβ were determined based on a zero tolerance. The results are presented in . CCα and CCβ were below the lowest validation level and thus below the recommended concentrations (CRL Citation2007) and/or RPAs (European Commission Citation2019). Therefore, the method is applicable for analysis of all these PPAS in bovine milk. The validation of CPZ showed a deviating trueness at two levels. However, CCβ could be established because the validation showed 21 (100%) non-compliant results at 0.5 times validation level. Therefore, CCβ is equal or below 0.5 µg kg−1. Consequently, for CPZ, the validation proved that the method is applicable for qualitative analysis of CPZ in bovine milk.
The CCα and CCβ results of NFs and CAP in the reported method are slightly higher compared to the validation results of NFs and CAP in milk published by Kaufmann et al. (Kaufmann et al. Citation2015) However, in this method only two substance classes (NFs and CAP) were included and in the method described in this study five different classes were included. In addition, the CCα and CCβ values described in this study are low enough to detect the PPAS at the required or recommended levels. On the other hand, the CCα and CCβ values presented in this study shows a strong improvement compared to the multimethod published by Kibechu et al. including NIZ, CAP, DAP and CPZ (NFs not included) and with LOQ values between 6 and 37 μg kg−1 (Kibechu and Sichilongo Citation2012)
Confirmation of identity
Following Commission Decision 2002/657/EC (European Commission Citation2002), the identity was confirmed for all substances, in all validation samples. Therefore, the validation proved that the method is applicable to confirm the identity of the PPAS at relevant levels.
It should be noted that confirmation of the identity of SEM does not automatically confirms the use of nitrofurazone at dairy farms although SEM is the mentioned marker metabolite in the guidelines (CRL Citation2007). The specificity of SEM as marker metabolite for nitrofurazone has been under debate for years (EFSA Citation2015; Abernethy Citation2015; Stadler et al. Citation2015). SEM has been detected despite no nitrofurazone being used, especially in processed milk products. SEM in dairy products might be a by-product which is produced during the manufacturing process. So far, SEM has not been detected in raw milk samples as a by-product and this allows us to use SEM as a marker metabolite for nitrofurazone in this method which is specific for raw milk samples (Stadler et al. Citation2015).
Stability
The stability was tested for the reference standards in solution and in the final extract. The NIZs, NFs (except DNSH) and DAP demonstrated to be stable in stock solution for at least one year and CAP for at least six months when stored in the fridge. CPZ is stable for at least two years stored in the freezer and DNSH is stable for at least one month in the freezer.
The stability in extracts was tested by re-analysing seven final extracts after 1.5 weeks storage in the freezer. For all substances, the trueness was between 96% and 108% and the RSDr* between 1.4% and 10.3% and both are acceptable: no relevant degradation is observed. Therefore, the final extracts are stable for at least one and a half weeks stored in the freezer.
Additional validation
The method was additionally validated for caprine milk, and the results are presented in . The selectivity of the method is considered sufficient because no interfering signals were observed in the blank samples. The trueness of all substances complied with the criteria except for MNZ and TNZ. The RSDr* complies for all substances and the RSDRL complies for all substances, except for MNZ-OH. Therefore, MNZ, TNZ and MNZ-OH cannot be analysed quantitatively, but can be analysed quali-tatively using the reported method. The CCα and CCβ could not be calculated, due to the limited number of data points. However, all substances were detected (with signal-to-noise ratio > 10) at validation level and thus it is concluded that CCβ is equal to or below the validation level. Consequently, CCα is in all cases below the validation level. The identity of all substances was confirmed in all cases and therefore the method is suitable for confirmation of the identity of the PPAS.
Table 3. The validation results in caprine milk; trueness, repeatability including matrix variance (RSDr*), within-laboratory reproducibility (RSDRL), decision limit (CCα), detection capability (CCβ). The underlined values exceed the established criteria
Validation performances
The reported method for the analysis of PPAS in milk is relative time-consuming. However, the complete validation showed that all PPAS of five different substance classes could be detected and confirmed in caprine and bovine milk at relevant levels within a single method. In addition, 13 out of 16 PPAS could also be quantified in caprine milk and 15 out of 16 PPAS could be quantified in bovine milk.
Supplemental Material
Download MS Word (126.6 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Abernethy GA. 2015. Generation of semicarbazide from natural azine development in foods, followed by reaction with urea compounds. Food Addit Contam Part A. 32(9):1416–1430. doi:https://doi.org/10.1080/19440049.2015.1067724.
- Aldeek F, Hsieh KC, Ugochukwu ON, Gerard G, Hammack W. 2018. Accurate quantitation and analysis of nitrofuran metabolites, chloramphenicol, and florfenicol in seafood by ultrahigh-performance liquid chromatography–tandem mass spectrometry: method validation and regulatory samples. J Agric Food Chem. 66(20):5018–5030. doi:https://doi.org/10.1021/acs.jafc.7b04360.
- Alkan F, Kotan A, Ozdemir N. 2016. Development and validation of confirmatory method for analysis of nitrofuran metabolites in milk, honey, poultry meat and fish by liquid chromatography-mass spectrometry. Maced Vet Rev. 39(1):15–22. doi:https://doi.org/10.1515/macvetrev-2015-0060.
- Allen EH. 1985. Review of chromatographic methods for chloramphenicol residues in milk, eggs, and tissues from food-producing animals. J AOAC Int. 68(5):990–999. doi:https://doi.org/10.1093/jaoac/68.5.990.
- Amelin VG, Fedina NM, Podkolzin IV, Korotkov AI. 2018. Rapid screening and determination of residual veterinary drugs in milk by ultrahigh performance liquid chromatography–high-resolution quadrupole time-of-flight mass spectrometry. J Anal Chem. 73(6):576–585. doi:https://doi.org/10.1134/S1061934818060023.
- Andries JPM, De Vries AB. 2007. Chemometrie. 3th ed. Arnhem (The Netherlands): Uitgeverij Syntax Media.
- Baynes RE, Dedonder K, Kissell L, Mzyk D, Marmulak T, Smith G, Tell L, Gehring R, Davis J, Riviere JE. 2016. Health concerns and management of select veterinary drug residues. Food Chem Toxicol. 88:112–122. doi:https://doi.org/10.1016/j.fct.2015.12.020.
- Berendsen BJA, Elbers IJW, Stolker AAM. 2011. Determination of the stability of antibiotics in matrix and reference solutions using a straightforward procedure applying mass spectrometric detection. Food Addit Contam Part A. 28(12):1657–1666.
- Berlina AN, Taranova NA, Zherdev AV, Vengerov YY, Dzantiev BB. 2013. Quantum dot-based lateral flow immunoassay for detection of chloramphenicol in milk. Anal Bioanal Chem. 405(14):4997–5000. doi:https://doi.org/10.1007/s00216-013-6876-3.
- Boechat N, Carvalho AS, Salomão K, De Castro SL, Araujo-Lima CF, Mello FVC, Felzenszwalb I, Aiub CAF, Conde TR, Zamith HPS, et al. 2015. Studies of genotoxicity and mutagenicity of nitroimidazoles: demystifying this critical relationship with the nitro group. Mem Inst Oswaldo Cruz. 110(4):492–499. doi:https://doi.org/10.1590/0074-02760140248.
- [CRL] Community Reference Laboratories’. 2007. CRL guidance paper (7 December 2007), CRLs View on state of the art analytical methods for national residue control plans.
- Chen D, Delmas J-M, Hurtaud-Pessel D, Verdon E. 2020. Development of a multi-class method to determine nitroimidazoles, nitrofurans, pharmacologically active dyes and chloramphenicol in aquaculture products by liquid chromatography-tandem mass spectrometry. Food Chem. 311:125924. doi:https://doi.org/10.1016/j.foodchem.2019.125924.
- [EMA] European Medicines Agency. 1996a. The European Agency for the evaluation of medicinal products veterinary medicines and information technology, Committee for veterinary medicinal products, Chlorpromazine, Summary Report. https://www.ema.europa.eu/documents/mrl-report/chlorpromazine-summary-report-committee-veterinary-medicinal-products_en.pdf.EMEA/MRL/111/96-FINAL .
- [EMA] European Medicines Agency. 1996b. The European Agency for the Evaluation of medicinal products veterinary medicines and information technology, Committee for veterinary medicinal products, Dapsone (2), Summary Report. https://www.ema.europa.eu/documents/mrl-report/dapsone-summary-report-2-committee-veterinary-medicinal-products_en.pdf .
- [EFSA] European Food Safety Authority. 2005. Opinion of the scientific panel on food additives, flavourings, processing aids and materials in contact with food (AFC) on a request related to a 9th list of substances for food contact materials, Question N° EFSA-Q-2004-071, EFSA-Q-2004-094, EFSA-Q-2003-214, EFSA-Q-2003-222. EFSA J. 248:1–16.
- [EMA] European Medicines Agency. 2012. CVMP assessment report under Article 30 (3)of Regulation (EC) No 726/2004, for dapsone as an impurity in veterinary medicinal products containing sulphamethoxazole or other sulphonamides. https://www.ema.europa.eu/documents/report/cvmp-assessment-report-under-article-303-regulation-ec-no-726/2004-dapsone-impurity-veterinary-medicinal-products-containing-sulphamethoxazole-other-sulphonamides_en.pdf.EMA/CVMP/392271/2012 .
- [EFSA] European Food Safety Authority. 2015. Scientific opinion on nitrofurans and their metabolites in food, EFSA panel on contaminants in the food chain (CONTAM). EFSA J. 13(6):4140. doi:https://doi.org/10.2903/j.efsa.2015.4139.
- Chu P-S, Lopez MI. 2007. Determination of nitrofuran residues in milk of dairy cows using liquid chromatography−tandem mass spectrometry. J Agric Food Chem. 55(6):2129–2135. doi:https://doi.org/10.1021/jf063391v.
- Cronly M, Behan P, Foley B, Malone E, Martin S, Doyle M, Regan L. 2010. Rapid multi-class multi-residue method for the confirmation of chloramphenicol and eleven nitroimidazoles in milk and honey by liquid chromatography-tandem mass spectrometry (LC-MS). Food Addit Contam Part A. 27(9):1233–1246. doi:https://doi.org/10.1080/19440049.2010.489579.
- European Commission. 1996. Council Directive 96/22/EC of 29 April 1996 concerning the prohibition on the use of stockfarming of certain substances having hormonal or thyrostatic action and of beta-agonistst and repealing Directives 81/602/EEC, 88/146/EEC and 88/299/EEC. Off J Eur Commun. L125:3–9.
- European Commission. 2002. Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results (2002/657/EC). Off J Eur Commun. L221:8–36.
- European Commission. 2010. Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. Off J Eur Union. L15:1–72.
- European Commission. 2017. Regulation (EU) 2017/625 of the European Parliament and of the Council of 15 March 2017 on official controls and other official activities performed to ensure the application of food and feed law, rules on animal health and welfare, plant health and plant protection products, amending Regulations (EC) No 999/2001, (EC) No 396/2005, (EC) No 1069/2009, (EC) No 1107/2009, (EU) No 1151/2012, (EU) No 652/2014, (EU) 2016/429 and (EU) 2016/2031 of the European Parliament and of the Council, Council Regulations (EC) No 1/2005 and (EC) No 1099/2009 and Council Directives 98/58/EC, 1999/74/EC, 2007/43/EC, 2008/119/EC and 2008/120/EC, and repealing Regulations (EC) No 854/2004 and (EC) No 882/2004 of the European Parliament and of the Council, Council Directives 89/608/EEC, 89/662/EEC, 90/425/EEC, 91/496/EEC, 96/23/EC, 96/93/EC and 97/78/EC and Council Decision 92/438/EEC (Official Controls Regulation). Off J Eur Union. L95:1–142.
- European Commission. 2019. Commission Regulation (EU) 2019/1871 of 7 November 2019 on reference points for action for non-allowed pharmacologically active substances present in food of animal origin and repealing Decision 2005/34/EC. Off J Eur Union. L289:41–46.
- Ferguson J, Baxter A, Young P, Kennedy G, Elliott C, Weigel S, Gatermann R, Ashwin H, Stead S, Sharman M. 2005. Detection of chloramphenicol and chloramphenicol glucuronide residues in poultry muscle, honey, prawn and milk using a surface plasmon resonance biosensor and Qflex® kit chloramphenicol. Anal Chim Acta. 529(1–2):109–113. doi:https://doi.org/10.1016/j.aca.2004.11.042.
- Galeano Dı́az T, Guiberteau Cabanillas A, Acedo Valenzuela MI, Correa CA, Salinas F. 1997. Determination of nitrofurantoin, furazolidone and furaltadone in milk by high-performance liquid chromatography with electrochemical detection. J Chromatogr A. 764(2):243–248. doi:https://doi.org/10.1016/S0021-9673(96)00899-0.
- Gui Y, Ni Y, Kokot S. 2011. Simultaneous determination of three 5-nitroimidazoles in foodstuffs by differential pulse voltammetry and chemometrics. Chin Chem Lett. 22(5):591–594. doi:https://doi.org/10.1016/j.cclet.2010.12.014.
- Hadjigeorgiou M, Papachrysostomou C, Theodorou Z, Kanari P, Constantinou S. 2009. Determination of dapsone in meat and milk by liquid chromatography tandem mass spectrometry. Anal Chim Acta. 637(1–2):220–224. doi:https://doi.org/10.1016/j.aca.2009.01.001.
- Hernández-Mesa M, Carbonell-Rozas L, Cruces-Blanco C, García-Campaña AM. 2017. A high-throughput UHPLC method for the analysis of 5-nitroimidazole residues in milk based on salting-out assisted liquid–liquid extraction. J Chromatogr B. 1068-1069:125–130. doi:https://doi.org/10.1016/j.jchromb.2017.10.016.
- Hernández-Mesa M, García-Campaña AM, Cruces-Blanco C. 2014. Novel solid phase extraction method for the analysis of 5-nitroimidazoles and metabolites in milk samples by capillary electrophoresis. Food Chem. 145:161–167. doi:https://doi.org/10.1016/j.foodchem.2013.08.002.
- Hernández-Mesa M, García-Campaña AM, Cruces-Blanco C. 2018. Development and validation of a QuEChERS method for the analysis of 5-nitroimidazole traces in infant milk-based samples by ultra-high performance liquid chromatography-tandem mass spectrometry. J Chromatogr A. 1562:36–46. doi:https://doi.org/10.1016/j.chroma.2018.05.050.
- Hernández-Mesa M, Lara FJ, Cruces-Blanco C, García-Campaña AM. 2015. Determination of 5-nitroimidazole residues in milk by capillary electrochromatography with packed C18 silica beds. Talanta. 144:542–550. doi:https://doi.org/10.1016/j.talanta.2015.06.049.
- [JECFA] Joint FAO/WHO Expert Committee on Food Additives - World Health Organization & Food and Agriculture Organization of the United Nations. 1991. Evaluation of certain veterinary drug residues in food: thirty-eighth report of the Joint FAO/WHO Expert Committee on Food Additives. meeting held in Geneva 22 to 31 January 1991.
- [JECFA] Joint FAO/WHO Expert Committee on Food Additives - World Health Organization & Food and Agriculture Organization of the United Nations. 2004. Evaluation of certain veterinary drug residues in food: sixty-second report of the Joint FAO/WHO Expert Committee on Food Additives. meeting held in Rome 4 to 12 February 2004; Rome.
- Horwitz W, Kamps LR, Boyer KW. 1980. Quality assurance in the analysis of foods and trace constituents. J Assoc Off Anal Chem. 63(6):1344–1354.
- Jadhav MR, Pudale A, Raut P, Utture S, Ahammed Shabeer TP, Banerjee K. 2019. A unified approach for high-throughput quantitative analysis of the residues of multi-class veterinary drugs and pesticides in bovine milk using LC-MS/MS and GC–MS/MS. Food Chem. 272:292–305. doi:https://doi.org/10.1016/j.foodchem.2018.08.033.
- Kaklamanos G, Theodoridis G. 2012. Determination of dapsone in muscle tissue and milk using high-performance liquid chromatography–tandem mass spectrometry. J Agric Food Chem. 60(1):29–35. doi:https://doi.org/10.1021/jf203701f.
- Kaufmann A, Butcher P, Maden K, Walker S, Widmer M. 2015. Determination of nitrofuran and chloramphenicol residues by high resolution mass spectrometry versus tandem quadrupole mass spectrometry. Anal Chim Acta. 862:41–52. doi:https://doi.org/10.1016/j.aca.2014.12.036.
- Kibechu R, Sichilongo K. 2012. A liquid chromatography mass spectrometry method for the determination of some pharmacologically active substances validated using milk as a matrix. J Liq Chromatogr Relat Technol. 35(12):1697–1711. doi:https://doi.org/10.1080/10826076.2011.621497.
- León N, Roca M, Igualada C, Martins CPB, Pastor A, Yusá V. 2012. Wide-range screening of banned veterinary drugs in urine by ultra high liquid chromatography coupled to high-resolution mass spectrometry. J Chromatogr A. 1258:55–65. doi:https://doi.org/10.1016/j.chroma.2012.08.031.
- Mitrowska K, Antczak M, Posyniak A. 2014. Confirmatory method for the determination of nitroimidazoles in milk by liquid chromatography - tandem mass spectrometry. Bull Vet Inst Pulawy. 58(4):581–587. doi:https://doi.org/10.2478/bvip-2014-0090.
- Mohamed R, Richoz-Payot J, Gremaud E, Mottier P, Yilmaz E, Tabet J-C, Guy PA. 2007. Advantages of molecularly imprinted polymers LC-ESI-MS/MS for the selective extraction and quantification of chloramphenicol in milk-based matrixes - comparison with a classical sample preparation. Anal Chem. 79(24):9557–9565. doi:https://doi.org/10.1021/ac7019859.
- Molognoni L, Daguer H, Hoff RB. 2021. Chapter 12 - analysis of nitrofurans residues in foods of animal origin. In: Galanakis CM, editor. Food toxicology and Forensics. Cambridge (MA): Academic Press; p. 379–419.
- Mulder PPJ, Zuidema T, Keestra NGM, Kooij PJF, Elbers IJW, van Rhijn JA. 2005. Determination of nifursol metabolites in poultry muscle and liver tissue - Development and validation of a confirmatory method. Analyst. 130(5):763–771.
- Nicolich RS, Werneck-Barroso E, Sípoli Marques MA. 2006. Food safety evaluation: detection and confirmation of chloramphenicol in milk by high performance liquid chromatography-tandem mass spectrometry. Anal Chim Acta. 565(1):97–102. doi:https://doi.org/10.1016/j.aca.2006.01.105.
- Nouws JFM, Vree TB, Holtkamp J, Baakman M, Driessens F, Guelen PJM. 1986. Pharmacokinetic, residue and irritation aspects of chloramphenicol sodium succinate and a chloramphenicol base formulation following intramuscular administration to ruminants. Vet Q. 8:224–232. doi:https://doi.org/10.1080/01652176.1986.9694046.
- Ohkubo T, Shimoyama R, Sugawara K. 1993. Determination of chlorpromazine in human breast milk and serum by high-performance liquid chromatography. J Chromatogr B. 614(2):328–332. doi:https://doi.org/10.1016/0378-4347(93)80326-Y.
- Ortelli D, Cognard E, Jan P, Edder P. 2009. Comprehensive fast multiresidue screening of 150 veterinary drugs in milk by ultra-performance liquid chromatography coupled to time of flight mass spectrometry. J Chromatogr B. 877(23):2363–2374. doi:https://doi.org/10.1016/j.jchromb.2009.03.006.
- Perez N, Gutierrez R, Noa M, Diaz G, Luna H, Escobar I, Munive Z. 2002. Liquid chromatographic determination of multiple sulfonamides, nitrofurans, and chloramphenicol residues in pasteurized milk. J AOAC Int. 85(1):20–24. doi:https://doi.org/10.1093/jaoac/85.1.20.
- Rezende DR, Fleury Filho N, Rocha GL. 2012. Simultaneous determination of chloramphenicol and florfenicol in liquid milk, milk powder and bovine muscle by LC–MS/MS. Food Addit Contam Part A. 29(4):559–570. doi:https://doi.org/10.1080/19440049.2011.641161.
- Robert C, Gillard N, Brasseur P-Y, Pierret G, Ralet N, Dubois M, Delahaut P. 2013. Rapid multi-residue and multi-class qualitative screening for veterinary drugs in foods of animal origin by UHPLC-MS/MS. Food Addit Contam Part A. 30(3):443–457. doi:https://doi.org/10.1080/19440049.2012.751632.
- Rodziewicz L. 2008. Determination of nitrofuran metabolites in milk by liquid chromatography–electrospray ionization tandem mass spectrometry. J Chromatogr B. 864(1–2):156–160. doi:https://doi.org/10.1016/j.jchromb.2008.01.008.
- Ryan JJ, Lee YC, Dupont JA, Charbonneau CF. 1975. A screening method for determining nitrofuran drug residues in animal tissues. J Assoc Off Anal Chem. 58(6):1227–1231.
- Shen J, Wang W, Xia X, Zhu J, Wu X, Wang S, Niu L, Ding S, Wang Z, Wang Z, et al. 2013. Determination of four nitrofuran metabolites and chloramphenicolin biological samples using enzyme-linked immunosorbent assay. Anal Lett. 46(9):1404–1418. doi:https://doi.org/10.1080/00032719.2012.762583.
- Śniegocki T, Giergiel M, Sell B, Posyniak A. 2018. New method of analysis of nitrofurans and nitrofuran metabolites in different biological matrices using UHPLC-MS/MS. J Vet Res. 62(2):161–166. doi:https://doi.org/10.2478/jvetres-2018-0025.
- Shendy AH, Al-Ghobashy MA, Gad Alla SA, Lotfy HM. 2016. Development and validation of a modified QuEChERS protocol coupled to LC–MS/MS for simultaneous determination of multi-class antibiotic residues in honey. Food Chem. 190:982–989. doi:https://doi.org/10.1016/j.foodchem.2015.06.048.
- Sørensen LK, Elbæk TH, Hansen H. 2003. Determination of chloramphenicol in bovine milk by liquid chromatography/tandem mass spectrometry. J AOAC Int. 86(4):703–706. doi:https://doi.org/10.1093/jaoac/86.4.703.
- Stadler RH, Verzegnassi L, Seefelder W, Racault L. 2015. Why semicarbazide (SEM) is not an appropriate marker for the usage of nitrofurazone on agricultural animals. Food Addit Contam Part A. 32(11):1842–1850. doi:https://doi.org/10.1080/19440049.2015.1086028.
- Suhren G, Heeschen W. 1993. Detection of eight sulphonamides and dapsone in milk by a liquid chromatographic method. Anal Chim Acta. 275(1–2):329–333. doi:https://doi.org/10.1016/0003-2670(93)80310-H.
- European Commission. 2005. Commission Decision of 11 January 2005 laying down harmonised standards for the testing for certain residues in products of animal origin imported from third countries (2005/34/EC). Off J Eur Commun. L16:61–63.
- Thompson CS, Traynor IM, Fodey TL, Crooks SRH. 2009. Improved screening method for the detection of a range of nitroimidazoles in various matrices by optical biosensor. Anal Chim Acta. 637(1–2):259–264. doi:https://doi.org/10.1016/j.aca.2008.09.040.
- Thompson M. 2000. Recent trends in inter-laboratory precision at ppb and sub-ppb concentrations in relation to fitness for purpose criteria in proficiency testing. Analyst. 125(3):385–386. doi:https://doi.org/10.1039/b000282h.
- Tölgyesi Á, Sharma VK, Fekete S, Fekete J, Simon A, Farkas S. 2012. Development of a rapid method for the determination and confirmation of nitroimidazoles in six matrices by fast liquid chromatography–tandem mass spectrometry. J Pharm Biomed Anal. 64-65:40–48. doi:https://doi.org/10.1016/j.jpba.2012.02.013.
- Tuzimski T, Rejczak T. 2017. A QuEChERS-based sample preparation method for the analysis of 5-nitroimidazoles in bovine milk by HPLC–DAD. J AOAC Int. 100(6):1671–1680. doi:https://doi.org/10.5740/jaoacint.17-0213.
- van Reeuwijk LP. 1998. Guidelines for quality management in soil and plant laboratories, Chapter 7 quality of analytical procedures. Rome (Italy): International soil reference and information centre, Food and Agriculture Organization of the United Nations.
- Van Rhijn JA, Lasaroms JJP, Berendsen BJA, Brinkman UAT. 2002. Liquid chromatographic–tandem mass spectrometric determination of selected sulphonamides in milk. J Chromatogr A. 960(1–2):121–133. doi:https://doi.org/10.1016/S0021-9673(02)00329-1.
- Varenina I, Bilandžić N, Kolanović BS, Đ B, Sedak M, Đokić M, Varga I. 2016. Validation of a liquid chromatography-tandem mass spectrometry method for the simultaneous determination of sulfonamides, trimethoprim and dapsone in muscle, egg, milk and honey. Food Addit Contam Part A. 33(4):656–667.
- [WHO] World Health Organization - International Agency for Research on Cancer. 1987. IARC monographs on the evaluation of carcinogenic risks to humans. - overall evaluation of carcinogenicity: an updating of IARC monographs Volumes 1 to 42. Lyon (France): International Agency for Research on Cancer (IARC).
- Wang Q, Wang C, Wu Q, Wang Z. 2019. Preparation of a magnetic nanoporous polymer for the fast and efficient extraction of 5-nitroimidazoles in milk. J Agric Food Chem. 67(41):11527–11535. doi:https://doi.org/10.1021/acs.jafc.9b03127.
- Wang Y, Li X, Zhang Z, Ding S, Jiang H, Li J, Shen J, Xia X. 2016. Simultaneous determination of nitroimidazoles, benzimidazoles, and chloramphenicol components in bovine milk by ultra-high performance liquid chromatography–tandem mass spectrometry. Food Chem. 192:280–287. doi:https://doi.org/10.1016/j.foodchem.2015.07.033.
- Xia X, Li X, Zhang S, Ding S, Jiang H, Li J, Shen J. 2008. Simultaneous determination of 5-nitroimidazoles and nitrofurans in pork by high-performance liquid chromatography–tandem mass spectrometry. J Chromatogr A. 1208(1–2):101–108. doi:https://doi.org/10.1016/j.chroma.2008.08.055.
- Zhan J, Yu X-J, Zhong -Y-Y, Zhang Z-T, Cui X-M, Peng J-F, Feng R, Liu X-T, Zhu Y. 2012. Generic and rapid determination of veterinary drug residues and other contaminants in raw milk by ultra performance liquid chromatography–tandem mass spectrometry. J Chromatogr B. 906:48–57. doi:https://doi.org/10.1016/j.jchromb.2012.08.018.
- Zhang Z, Wu Y, Li X, Wang Y, Li H, Fu Q, Shan Y, Liu T, Xia X. 2017. Multi-class method for the determination of nitroimidazoles, nitrofurans, and chloramphenicol in chicken muscle and egg by dispersive-solid phase extraction and ultra-high performance liquid chromatography-tandem mass spectrometry. Food Chem. 217:182–190. doi:https://doi.org/10.1016/j.foodchem.2016.08.097.