
ABSTRACT
The bacterial communities of Zaopocu (ZPC), a tropical specialty fermented food in Hainan Island, were characterized in this study using high-throughput sequencing. Firmicutes and Proteobacteria were the two dominant phyla. The primary genus in ZPC_A was Weissella, while ZPC_B and ZPC_C had Lactobacillus as the dominant genus. The biomarker lactic acid bacteria (LAB) in ZPC_A were Lactobacillus brevis, Weissella confusa, and Leuconostoc citreum, whereas Lactobacillus amylolyticus and Lactobacillus helveticus were significantly higher in ZPC_B. In contrast, Lactobacillus pontis and Lactobacillus fermentum were the core LAB in ZPC_C. ZPC_A has the highest value of titratable acid (TA), amino acid, and lactic acid. TA had the most powerful influence on the structure of the bacterial communities, whereas salinity had a negligible effect. It is worth noting that ZPC may also faces food safety issue because opportunistic pathogenic bacteria like Stenotrophomonas and genes closely associated with Staphylococcus aureus infection were highly detected.
1. Introduction
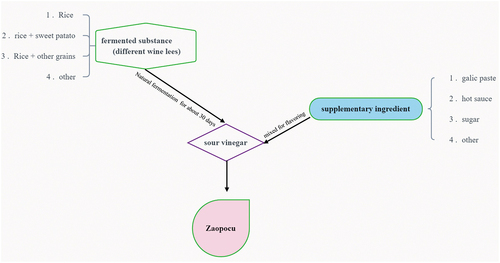
As the name implies, Zaopocu (ZPC) is sour vinegar produced by further fermentation of the wine lees. The manufacture of Chinese rice wine has become widely known. First, different grains, mainly rice, are steamed and cooled, then fermentation begins by adding different Qu (starter) and mixing it with the additional water (Cai et al., Citation2018). Then the lees were sealed, settled, and fermented for about one month, and the acidic liquid was finally harvested by filtering the residue. Additional ingredients such as garlic, salt, and hot sauce are subsequently added into sour vinegar for further processing and flavoring. So actually, ZPC is made up of acidic vinegar and various additives (). Moreover, due to the different fermentation substrates and additives used by producers, ZPC can have multiple flavors, ranging from sour to sweet to spicy. As a result, ZPC produces a unique and fantastic taste that sets it apart from other Hainan Island fermented foods.
Figure 1. The manufacturing process of Zaopocu.
The lactic acid bacteria (LAB) is a heterogeneous group with these common features: catalase negativity, non-spore formation, fermentative, facultative aerobic, and producing lactic acid by fermented glucose (Salvetti et al., Citation2012). LAB can be readily found in plants, and mammal intestinal mucosa, especially in fermented foods, where they contribute significantly to the fermented food’s flavors, sensory taste, and nutritional value (Parvez et al., Citation2006). Different types of Chinese fermented foods, such as Suancai (He et al., Citation2020), yogurt (Liang et al., Citation2021), and Douchi (Y. Z. Zhang et al., Citation2022), vary significantly in LAB diversity, and providing a potential resource trove of probiotics. The traditional culture-dependent method starts with screening culturable microorganisms via culture medium, followed by microscopic, physiochemical, and molecular biology tests, which are essential for isolating and identifying LAB. Using the culture-dependent method, a total of 39 LAB strains were isolated from Pickled Cowpea (Z. Guo et al., Citation2021), a popular fermented vegetable in China. They were identified as Lactobacillus plantarum, Lactobacillus fermentum, and Lactobacillus rhamnosus. While in JiangShui (Jun et al., Citation2018), the isolated LAB strains are closely related to Lactobacillus brevis, Lactobacillus plantarum, and Lactobacillus parabuchneri. However, traditional culture-based methods can only isolate some culturable microorganisms, and the number of these microorganisms is negligible compared to those that cannot be grown on culture media. High-throughput sequencing (HTS) can identify microbial species that cannot be grown using traditional culture methods, especially in samples with complex microbial communities. With the ability to provide more precise information on the bacterial composition, HTS holds significant potential in exploring the diversity of LAB in fermented foods (Lemos et al., Citation2011). With the help of HTS, the related studies found that LAB composition may differ significantly even in the same type of fermented food. For example, Lactobacillus pontis, Lactobacillus amylolyticus, and Lactobacillus fermentum dominate in Jiang-Shui (Northwest China) samples. In contrast, Lactobacillus coryniformis, Lactobacillus pentosus, and Lactobacillus parabrevis are predominant in the Suan-cai (Southern China) samples (Liu, Li, et al., Citation2019).
Hainan Island is located at the southernmost of China and has unique climatic conditions. Recently researchers have reported various fermented foods with individual microbial communities and unique LAB compositions, such as Yucha (J. Zhang et al., Citation2016) and tropical fermented fruits and vegetables (Peng et al., Citation2018). These results contribute to the study of microbial diversity in Chinese fermented foods with Hainan characteristics. However, As far as we know, no study has examined ZPC’s microbial diversity. This study aimed to elucidate the composition of bacterial communities and the LAB diversity in three types of ZPC by high-throughput sequencing based on 16s DNA (V3-V4). We also propose to identify some core bacteria that are closely linked to physicochemical factors and thus try to explore the important role that these specific bacteria may play in ZPC production. This work may enhance the understanding of the microbial communities in ZPC for the first time, probing the LAB resources in ZPC, thus providing the basis for the future development of ZPC-derived probiotics.
2. Materials and methods
2.1. ZPC sample collection
A total of 12 independent ZPC samples were collected from three local markets (4 separate samples from different manufacturers) in Haikou, Hainan province. The collected samples were sorted into three main categories, with the following distinctions: the fermentation substrate of ZPC_A came from yellow wine, while ZPC_B and ZPC_C were from rice wine. In addition, a small amount of hot sauce was added to ZPC_A, and ZPC_B contained a little garlic paste, while ZPC_A had no added substances (). The samples were then divided into small portions and frozen immediately at −80°C for DNA extraction or stored at 4°C for physicochemical index analysis.
Table 1. The characteristics of collected three types of Zaopocu samples.
2.2. Basic physicochemical parameters analysis
A pH meter (METTLER TOLEDO FE28) and a salinity meter (Qingdao Tlead, SA287) were used to detect the value of pH and salinity. The potentiometric titration method was used (0.01 mol/L sodium hydroxide) to determine the titratable acid (TA, g/L) in ZPCs following the national standard -Determination of total acid in Foods (GB-12456–2021). The amount of NaOH used was recorded when the titration endpoint reached 8.2 ± 0.2. The coefficient of lactic acid (0.09) was then used to calculate the final TA value. Meanwhile, various biochemical assay kits (Boxbio, Beijing) were used to determine the content of nitrite, reducing sugars, and free amino acids (AA) at 540, 540, and 570 nm, respectively. Lactic acid (LA) concentrations were measured using the HPLC method as previously described (Degrain et al., Citation2020).
2.3. DNA extraction and PCR amplification
In order to extract genomic DNA from ZPC samples, we used Tiangen Fecal Genomic DNA Extraction Kit (Tiangen, Beijing). The concentration and quality of DNA were determined via electrophoresis on 1% agarose gels. The bacterial 16S rDNA V3-V4 region was amplified using primer pair 338F (5”ACTCCTACGGGAGGCAGCAG-3”) and 806 R (5”-GGACTACHVGGGTWTCTAAT-3”) (Poirier et al., Citation2018). PCR reaction conditions included: After predenaturation at 95°C for 5 minutes, 30 cycles of denaturing at 95°C for 30 seconds, annealing at 60°C for 30 seconds, extension at 72°C for 1 minute, and final extension at 72°C for 7 minutes, the sample was extended for 7 minutes. PCR reactions were performed in a 50 μL mixture containing 10 μL of 5 × Q5@ Reaction Buffer, 10 μL of 5 × Q5@ High GC Enhancer, 1.5 μL of 2.5 mM dNTPs, 1.5 μL of each primer (10 μM), 0.2 μL of Q5@ High-Fidelity DNA Polymerase, and 50 ng of template DNA. A complete set of PCR reagents was obtained from New England Biolabs, U.S.A.
2.4. Illumina sequencing and bioinformatics analysis
The amplicons were extracted from 2% agarose gels using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, U.S.). The amplicons were then quantified using ABI StepOnePlus Real-Time PCR System (Life Technologies, U.S.A). ABI StepOnePlus Real-Time PCR System was used to quantify the amplicons, after which the purified amplicons were pooled in equimolar quantities and sequenced with an Illumina platform at Gene Denovo Bio (Guangzhou city, China).
FASTP was used to filter raw reads, and then FLASH was used to merge the paired-end clean reads into raw tags. UPARSE pipeline was used to cluster clean tags into operational taxonomic units (OTUs) of 97% similarity (Edgar, Citation2013). The UCHIME algorithm was used to remove all chimeric tags (Edgar et al., Citation2011). The obtained effective tags were used for further analysis. The following alpha-diversity indexes were all calculated using QIIME (Caporaso et al., Citation2010): Chao1, ACE, Shannon, Simpson, Good’s coverage, and Pielou’s evenness. The R project Vegan package was used for principal component analysis (PCA), heatmap analysis, and the UpSet Venn plot. LEfSe software was used to screen the biomarker features in each group. Based on the OTU level, KEGG pathway analysis was performed using PICRUSt (Douglas et al., Citation2018). BugBase was used to classify bacterial microbiome phenotypes (Ward et al., Citation2017).
2.5. Statistical analysis
Data are expressed as mean ± Standard Error (SE). Welch’s t-test was employed to analyze the differences in physicochemical characteristics and microbial community phenotypes. Tukey’s HSD test was used to compare alpha indices and the metabolic pathways difference among the three ZPC groups, P-value < .05 was considered significant. Canonical Correspondence Analysis (CCA) and Mantel tests were conducted to investigate the correlation between bacterial communities and physicochemical indicators.
3. Materials and methods
3.1. Physicochemical properties of ZPC samples
The pH, salinity, and nitrite values were not significantly different among the three groups (). However, ZPC_A and ZPC_B have slightly higher pH and salinity than ZPC_C (P > .05). The nitrite content in all samples were below the national food safety standard- Determination of Nitrite and Nitrate in Foods (0.2 mg/kg, GB/T 5009.33–2010). Reducing sugar was not detected in ZPC_A and the concentration did not show a significant difference between ZPC_B (3.21) and ZPC_C (3.06). The highest value of TA, LA, and AA all existed in ZPC_A samples (P < .01) ().
Figure 2. The physicochemical properties of the three types of ZPC. (a) TA. (b) Lactic acid content. (c) Amino Acid content. (d) Nitrite content. (e) pH. (f) Salinity. (g) Reducing sugar content. *P < .05, **P < .01, ***P < .001, ns= no significant.
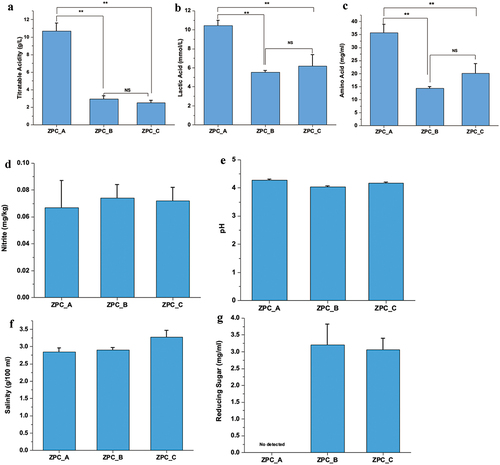
Table 2. The physicochemical properties of 12 ZPC samples.
3.2. Sequencing data characteristics and α-diversity indices
A brief summary of the sequence data information and the α-diversity index is shown in . A total of 752,566 sequences (average length; about 460 bp) and 2227 OTUs were acquired by filtering low-quality sequences and then classified at 97% similarity. ZPC_A, ZPC_B, and ZPC_C had 104, 223, and 229 OTUs, respectively. All samples had a good coverage index > 0.99, indicating that the sequencing was sufficient and covered most bacteria phylotypes. More observed species were detected as the sequencing progressed (). The rarefaction curve leveled off when the sequencing depth was over 20,000, indicating that the obtained OTUs could reflect the microbial richness and diversity of ZPC samples. Significant variations were observed in the four alpha-diversity indices of Shannon, Chao1, ACE, and Simpson. The ZPC_B samples had the highest Simpson and Shannon indices (), followed by ZPC_C and ZPC_A, and there was a noticeable difference among the three groups (P < .001). The Chao1 and ACE indexes were highest in the ZPC_C () and lowest in ZPC_A samples (P > .05). These results indicate that ZPC_B samples have the highest bacteria diversity, while the most outstanding bacterial richness is presented in the ZPC_C group.
Figure 3. Rarefaction curves of observed species number.
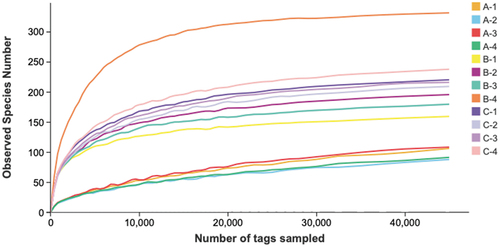
Figure 4. Comparison of alpha diversity indices among the three types of ZPC. (a) Shannon. (b) Simpson. (c) ACE. (d) Chao 1.*P < .05, **P < .01, ***P < .001, “ns” = not significant.
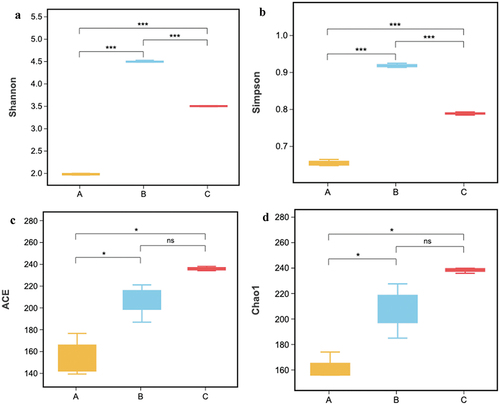
Table 3. Sequence data information and alpha diversity index of 12 ZPC samples.
3.3. Bacterial community diversity
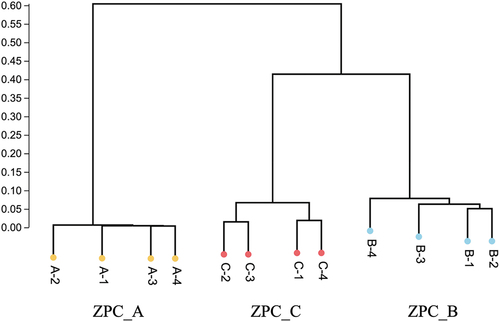
A total of 18 phyla, 200 genera, and 101 species were identified in all the samples (data not shown). ZPC_A had the lowest number of phyla (), in which Firmicutes (99.09%) solely dominated. Proteobacteria and Firmicutes dominate ZPC_B and ZPC_C, with Proteobacteria accounting for 56.52%, 48.40%, and Firmicutes for 35.29% and 46.18%. Furthermore, some phyla were detected only in specific groups. ZPC_B samples have the highest abundances of Cyanobacteria (6.29%), while the top content of Bacteroidetes (4.18%) was only detected ZPC_C group. ZPC_A basically consists of only three genera (), with Weissella dominating (48.88%), followed by Leuconostoc (39.45%) and Lactobacillus (10%). ZPC_C was dominated by Lactobacillus (43.13%) and Stenotrophomonas (26.51%). Lactobacillus (23.45%) was the sole dominant genus in ZPC_B samples, while the other genera were all below 10%, with Acetobacter as the second-ranked genus (8.11%). The composition at the species level () largely follows the genus level structure. ZPC_A and ZPC_C contained dominant bacteria, while ZPC_B had a relatively homogeneous structural composition, in which no species comprised more than 10% abundance. Weissella confusa (48.88%) was dominant in ZPC_A, followed by Leuconostoc citreum (30.77%). The two dominant species in ZPC_C samples were Lactobacillus fermentum (36.67%) and Stenotrophomonas maltophilia (26.47%). The highest abundances of Lactobacillus brevis (5.12%) and Lactobacillus pontis (2.96%) were detected in ZPC_A and ZPC_C, respectively. ZPC_B samples contained the highest abundances of Lactobacillus helveticus (2.28%) and Lactobacillus amylolyticus (8.37%).
Figure 5. Relative abundance (Top 10) of bacteria among the three types of ZPC samples at phylum (a,b), genus (c,d), and species (e,f) level.
3.4. Bacterial community comparison
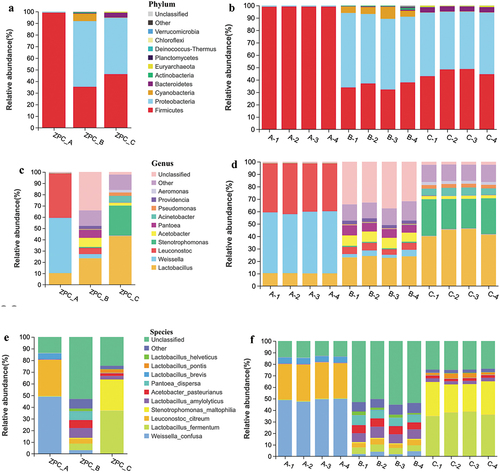
The three ZPC groups shared 63 identical OTUs (). However, the distribution of unique OTUs differed significantly among the three groups. ZPC_B and ZPC_C contained 252 and 127 unique OTUs, respectively. However, ZPC_A had only one special OTU belonging to the Weissella genus, the Leuconostocaceae family. Two-by-two analysis showed that ZPC_A and ZPC_C shared only two OTUs (), indicating that they have the lowest similarity in bacterial structural composition. PCA analysis showed the samples were clustered in three different areas at the OTU level, highlighting the distinct differences in bacteria composition among three types of ZPC (). ZPC_A samples were concentrated on the left side, while ZPC_C samples were focused on the far-right side, with 73.85% variance at the first axis, suggesting that the bacterial communities between them are relatively the most distinct. This result is further confirmed by statistical tests (p = .026).
Figure 6. Venn diagram (a) and Upset plots (b) for the shared/unique OTUs information.
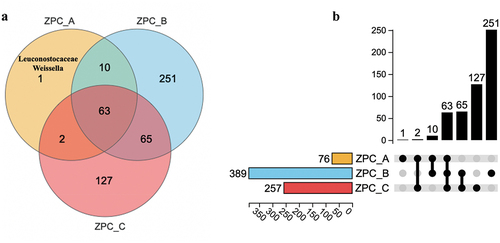
Figure 7. PCA analysis of the bacteria communities of the three types of ZPC samples based on OTU level.
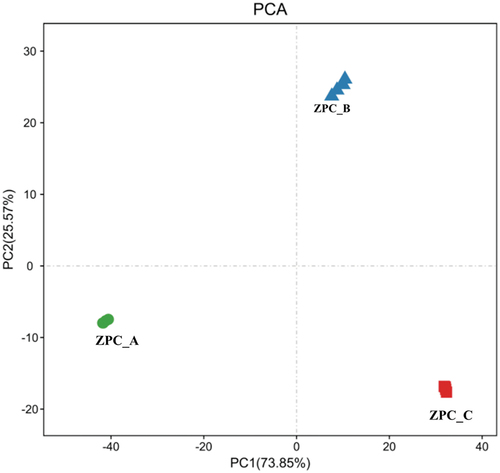
Two apparently independent clusters were observed in the UPGMA tree (), with the first group consisting of four samples (ZPC_A1- ZPC_A4); the remaining eight samples (ZPC_B1- ZPC_C4) formed the second group. The UPGMA analysis revealed a distinct clustering pattern among three types of ZPC (P = .0001) and further suggested that ZPC_A possesses significantly differing β-diversity.
Figure 8. Comparison of differences and similarity of the bacterial community structures among three types of ZPC samples by Cluster-tree. The clustering map was based on OTUs pattern and weighted_Unifrac distance.
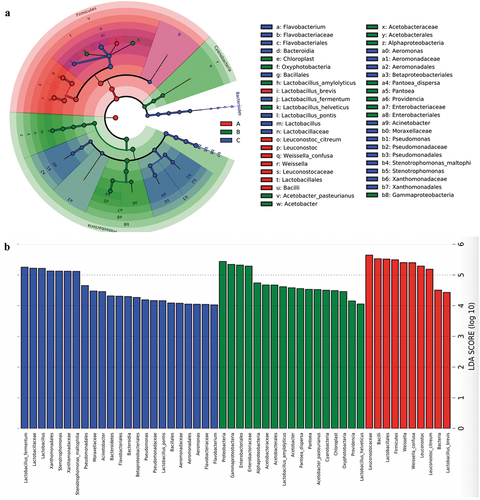
The linear discriminant analysis effect size (LEfSe) analysis showed a variety of core bacteria (biomarkers) among groups (). At the species level, Acetobacter pasteurianus, Lactobacillus amylolyticus, Pantoea dispersa, and Lactobacillus helveticus were enriched considerably in ZPC_B. Lactobacillus brevis, Weissella confusa, and Leuconostoc citreum were significantly enriched in ZPC_A, In contrast, Lactobacillus pontis,Lactobacillus fermentum, and Stenotrophomonas maltophilia were significantly enriched in ZPC_C samples.
Figure 9. LEfSe analyses the biomarker among the three types ZPCs. (a) The cladogram shows the abundance of different taxa in ZPC samples. (b) Histogram results with a threshold ≥ 4.
3.5. Predicted metabolic functions of the bacterial communities
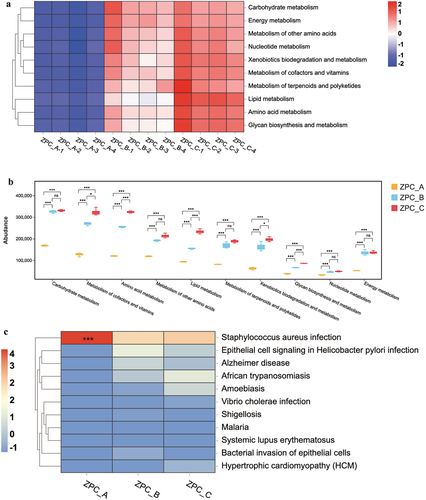
The PICRUSt predicted that the biological functions of bacterial communities in ZPC samples were enriched in six pathways: metabolism, genetic information processing, cellular processes, environmental information processing, organismal systems, and human disease (data not shown). The genes were enriched in 31 KEGG pathways based on level 2 analysis (), of which 16 were related to substance metabolism. ZPC_C had more significant bacterial activities related to metabolic function, especially the metabolism of carbohydrates, cofactors and vitamins, amino acids, and lipids (, P < .001). Other pathway functions were lower in the three groups and were not significantly different. Moreover, the level 3 () analysis detected 11 pathways related to human disease. Notably, the likelihood of S. aureus infection was remarkably high in all three ZPC groups, with ZPC_A samples exhibiting the highest probability (P < .001).
Figure 10. Bacterial function prediction by PICRUSt. (a) The heatmap analysis of the 10 substance metabolism pathways at level 2. (b) Statistical analysis of the metabolic pathways by the Turkey HSD test. *P < .05, ***P < .001, “ns”= not significant. (c) the heatmap analysis (level 3) of the functional pathways associated with human disease. ***P < .001 when compared with ZPC_B and ZPC_C.
3.6. Predictions of bacterial community function
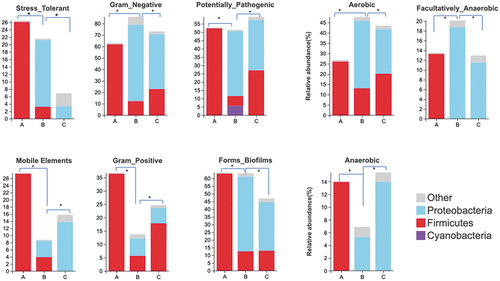
The Bug base predicted that aerobic and gram-negative bacteria were dominant in the three groups, with the highest levels detected in the ZPC_B (). Microbiota from ZPC_A samples contained the most abundant mobile elements, mainly originating from Firmicutes. Moreover, Proteobacteria provided all the mobile elements in ZPC_C samples. Biofilm-forming bacteria and potentially pathogenic bacteria (greater than 60%) were detected in all three groups with a similar origin. Notably, the three main phyla Firmicutes, Proteobacteria, and Cyanobacteria in the three groups were all potential sources of pathogenic bacteria in ZPC_B. Firmicutes were the dominant stress-tolerant bacteria in ZPC_A. However, the abundance of stress-tolerant bacteria was lowest in ZPC_C, mainly from Proteobacteria.
Figure 11. Prediction of microbial community phenotypes using the BugBase at the phylum level among three ZPC groups. *P < .05.
3.7. Relationship between physicochemical indexes and microbial communities
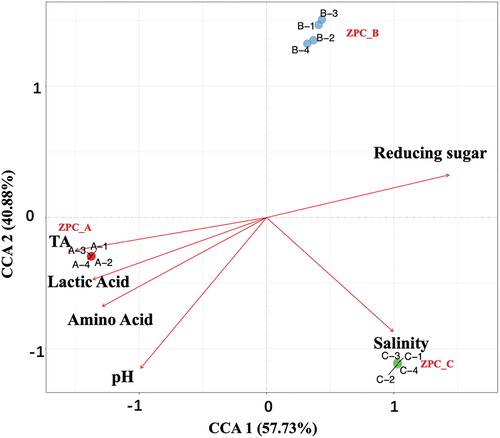
The canonical correlation analysis (CCA) clearly showed that physicochemical factors significantly influenced the composition of bacterial communities (). Mantel test results showed that TA (R2 = 0.9753, p = .002) had the most effect, followed by pH (R2 = 0.9751, p = .008). However, salinity had a negligible effect on the bacterial community (R2 = 0.6489, p = .08). In addition, the bacterial communities in ZPC_ B were positively correlated with the reducing sugar content, while salinity exhibited a positive association with the bacterial communities of ZPC_C. In contrast, TA and LA together shape the bacterial communities structure of ZPC_A.
Figure 12. Canonical correlation analysis (CCA) of three types of ZPC samples.
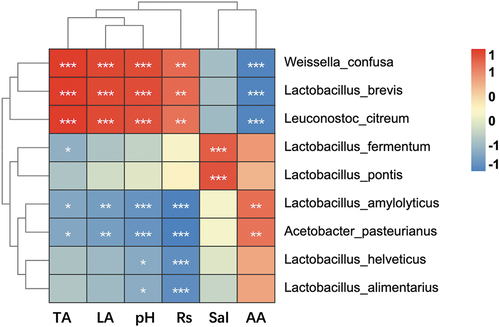
The Spearman correlation heatmap analysis () demonstrated some specific genera, especially the Lactobacillus. spp was significantly correlated with environmental factors. The abundances of Weissella confusa, Lactobacillus brevis, and Leuconostoc citreum were significantly positively correlated with TA, LA, pH, and reducing sugar; while showing a negatively correlated with AA content. The abundances of Lactobacillus fermentum and Lactobacillus pontis were positively correlated with salinity. In contrast, Lactobacillus amylolyticus and Acetobacter pasteurianus were positively correlated with AA content.
Figure 13. Spearman correlations analysis of the specific LAB and physicochemical characteristics. Red and blue represent positive and negative correlations, respectively. *P < .05, **P < .01, ***P < . 001.Rs = Reducing sugar. Sal = salinity.
4. Discussion
Due to the tropical climate, unique raw materials, and fermentation processes, fermented foods with Hainan Island characteristics have been reported some specificity microbial community structure and LAB diversity from other fermented foods on the mainland. However, studies on the microbial communities of ZPC have yet to be available, so Illumina MiSeq sequencing technology was first employed in this study to analyze the bacteria composition structure of three types of ZPCs. The most predominant bacterium present in ZPC_B and ZPC_C samples was Lactobacillus, which is consistent with the findings of reported Hainan fermented foods (Hu et al., Citation2020), and sour soup-a special fermented food from southwest China that is somewhat similar to the ZPC fermentation style (Lin et al., Citation2020). The LEFse analysis showed that Lactobacillus amylolyticus and Lactobacillus helveticus were dominant in ZPC_B, while Lactobacillus pontis and Lactobacillus fermentum were dominant in ZPC_C. However, only Lactobacillus brevis was dominant in ZPC_A, indicating Lactobacillus. spp composition greatly varied among the ZPC groups.In overall comparison, Lactobacillus sakei and Lactobacillus saniviri are the LAB in Yucha (J. Zhang et al., Citation2016), while Lactobacillus plantarum, Lactobacillus fermentum, Lactobacillus pentosaceus are dominant in various Hainan fermented fruits and vegetables (Peng et al., Citation2018). Meanwhile, Lactobacillus acetotolerans was the extremely dominant species in Red sour soup. These contrasting findings further suggest that the LAB diversity varies significantly even among fermented foods from the same region and with similar fermentation style.
The sour soup is fermented similarly to ZPC by co-fermenting fresh chilies and tomatoes while producing rice wine and Lactobacillus rhamnosus was identified to have a positive correlation with the formation of flavor substances in red sour soup (Zhou et al., Citation2022), indicating that LAB not only can prevent the spoilage of fermented foods but also play an essential role in developing the flavor substances in fermented foods. Our study confirmed Lactobacillus as the dominant genus in ZPC, suggesting their crucial role in the production of ZPC. But an obvious shortcoming of this study is that no systematic analysis of metabolites in ZPC was performed.
Therefore, the flavor formation of ZPC is closely related to which core LAB remains to be uncovered by future studies.
Notably, alpha and beta diversity analysis showed that the bacterial communities in ZPC_A were significantly different from ZPC_B and ZPC_C samples. Weissella (48.96%) and Leuconostoc (39.45%) almost equally divided the composition, while the abundance of Lactobacillus (10.28%) was just over 10%. This differs from the findings in sour soups, where Lactobacillus are extremely dominant, accounting for over 95% of the bacteria composition. Weissella (Dey et al., Citation2019) and Leuconostoc (Cho et al., Citation2006) genera have recently attracted much attention as new model strains of LAB. To our knowledge, the high abundance of these two bacteria has been detected for the first time in Hainan tropical fermented foods, suggesting that ZPC may have more different types of LAB than other Hainan fermented foods (Jiang et al., Citation2019; J. Zhang et al., Citation2016). In addition, we inferred that the high abundance of Weissella confusa and Leuconostoc citreum in ZPC_A is the cause of its unique fermentation substrate. In contrast to traditional single rice wine, yellow wine is produced by the co-fermentation of various mixed grains, including millet, rice, sorghum, or others. Related studies have also provided some valuable clues to our speculation; the high levels of these two bacteria have also been detected in single ferments, such as millet wine (Yan et al., Citation2022) and black glutinous rice wine (Zhao et al., Citation2020) in China, suggesting that multiple grains can bring more types of LAB during yellow rice wine fermentation. Therefore, ZPC_A naturally inherits these two genera from the yellow wine lees.
CCA analysis showed that TA significantly impacted the structure of the ZPC bacterial community, consistent with most studies on fermented foods in China (Liu, Peng, et al., Citation2019). A high TA value is often associated with the accumulation of lactic acid and other acids in a fermented vegetable product (Xiong et al., Citation2012). The same results were also observed in our study, as both TA and LA levels were highest in the ZPC_A samples. We inferred that this result may possibly be due to the high abundance of Weissella and Leuconostoc bacteria in the ZPC_A group. Because Weissella and Leuconostoc are found to be related to the accumulation of substance content in fermented foods, such as organic acids (Teixeira et al., Citation2021) and short-chain fatty acids (Yu et al., Citation2019). Therefore, Spearman’s analysis result is to be expected that the abundance of Weissella confusa and Leuconostoc citreum were significantly positively correlated with TA and LA. However, salinity had little effect on the ZPC’s bacterial communities, inconsistent with most research findings (Kim et al., Citation2020; Lee et al., Citation2021). A certain amount of salt is essential for preventing food spoilage, so the salt will powerfully shape the microbial communities during food fermentation. However, the fermentation substrate of ZPC is mainly lees produced in the rice wine brewing process. Because salt is not usually added during traditional Chinese rice wine production, thus ZPC has no salt involved in fermentation until the acetic acid is produced. Only a tiny amount of salt is added when additional substances are involved for flavoring. By this time, the bacterial structure has stabilized, and therefore, the effect of salinity on the bacterial composition of the ZPC is very slight.
Interestingly, in the ZPC_C group with the highest salinity values, we found that the abundance of Lactobacillus fermentum and Lactobacillus pontis were significantly positively correlated with salinity. Resistance to salt stress has always been a prerequisite for screening probiotics because they must tolerate high concentrations of bile salts in the body to achieve their efficacy (X. H. Guo et al., Citation2010). LAB bacteria isolated from certain high-salt fermented foods, such as Chinese fermented soybean paste (Li et al., Citation2015) and jet-gal- fermented seafood from Thailand (Song et al., Citation2021) have demonstrated that they possess significant probiotic efficacy. Whether these two bacteria have considerable resistance to salt stress and thus have the potential to become probiotics remains to be confirmed by subsequent isolations and in vitro/in vivo experiments.
It is worth mentioning that the content of Stenotrophomonas in the ZPC_C group is remarkably high, close to one-third of the entire bacterial composition. Although it is also frequently detected in some fermented foods, its presence is typically at low levels.For example, in two Chinese traditionally fermented vegetables (Guan et al., Citation2020), Suancai and Suansun, the percentage of Stenotrophomonas was only 6.33% and 1.74%, respectively. Stenotrophomonas maltophilia is an emerging global opportunistic pathogen reported to cause common infections such as urinary tract and pneumonia infections. It is widely present in the environment and has been recovered from soils and plant roots (Brooke, Citation2012). Of the three types of ZPC, only the lees of ZPC_C contain the additional material-sweet potato, so we assumed that the high levels of Stenotrophomonas are probably due to insufficient sterilization conditions in ZPC_C fermentation. Thus the wine lees were contaminated by Stenotrophomonas on the surface of sweet potatoes. Microbial safety risk caused by microbial contamination and various toxins in fermented foods has attracted much research attention (Anal et al., Citation2020). Therefore, appropriate measures are needed to prevent contamination of ZPC by opportunistic pathogens and other microorganisms.
In the KEGG analysis, genes closely associated with human infectious diseases were identified, with those associated with Staphylococcus aureus being enriched in all three groups. S. aureus is the most common contaminating microorganism in food (Huang et al., Citation2021), implying that higher hygiene standards are required during ZPC preparation and storage. Notably, a high proportion of potentially pathogenic and biofilm-forming bacteria was also predicted in bacterial community function analysis. Therefore, pathogenic bacteria harboring genes that can cause other infections should be explored in future studies.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets analyzed for High-throughput sequencing during the current study are available on a online platform (http://www.omicsmart.com), Details of access can be obtained from the corresponding author upon reasonable request.
Additional information
Funding
References
- Anal, A. K., Perpetuini, G., Petchkongkaew, A., Tan, R., Avallone, S., Tofalo, R., Van Nguyen, H., Chu-Ky, S., Ho, P. H., & Phan, T. T. (2020). Food safety risks in traditional fermented food from South-East Asia. Food Control, 109, 106922. https://doi.org/10.1016/j.foodcont.2019.106922
- Brooke, J. S. (2012). Stenotrophomonas maltophilia: An emerging global opportunistic pathogen. Clinical Microbiology Reviews, 25(1), 2–41. https://doi.org/10.1128/CMR.00019-11
- Cai, H., Zhang, T., Zhang, Q., Luo, J., Cai, C., & Mao, J. (2018). Microbial diversity and chemical analysis of the starters used in traditional Chinese sweet rice wine. Food Microbiology, 73, 319–326. https://doi.org/10.1016/j.fm.2018.02.002
- Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., Fierer, N., Peña, A. G., Goodrich, J. K., & Gordon, J. I. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7(5), 335–336. https://doi.org/10.1038/nmeth.f.303
- Cho, J., Lee, D., Yang, C., Jeon, J., Kim, J., & Han, H. (2006). Microbial population dynamics of kimchi, a fermented cabbage product. FEMS Microbiology Letters, 257(2), 262–267. https://doi.org/10.1111/j.1574-6968.2006.00186.x
- Degrain, A., Manhivi, V., Remize, F., Garcia, C., & Sivakumar, D. (2020). Effect of lactic acid fermentation on color, phenolic compounds and antioxidant activity in African nightshade. Microorganisms, 8(9), 1324. https://doi.org/10.3390/microorganisms8091324
- Dey, D. K., Koo, B. G., Sharma, C., & Kang, S. C. (2019). Characterization of Weissella confusa DD_A7 isolated from kimchi. LWT, 111, 663–672. https://doi.org/10.1016/j.lwt.2019.05.089
- Douglas, G. M., Beiko, R. G., & Langille, M. G. (2018). Predicting the functional potential of the microbiome from marker genes using PICRUSt. Microbiome Analysis: Method and Protocols, 169–177. https://doi.org/10.1007/978-1-4939-8728-3_11
- Edgar, R. C. (2013). UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10(10), 996–998. https://doi.org/10.1038/nmeth.2604
- Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., & Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27(16), 2194–2200. https://doi.org/10.1093/bioinformatics/btr381
- Guan, Q., Zheng, W., Huang, T., Xiao, Y., Liu, Z., Peng, Z., Gong, D., Xie, M., & Xiong, T. (2020). Comparison of microbial communities and physiochemical characteristics of two traditionally fermented vegetables. Food Research International, 128, 108755. https://doi.org/10.1016/j.foodres.2019.108755
- Guo, X. H., Kim, J. M., Nam, H. M., Park, S. Y., & Kim, J. M. (2010). Screening lactic acid bacteria from swine origins for multistrain probiotics based on in vitro functional properties. Anaerobe, 16(4), 321–326. https://doi.org/10.1016/j.anaerobe.2010.03.006
- Guo, Z., Wang, Y., Xiang, F., Hou, Q., & Zhang, Z. (2021). Bacterial diversity in pickled cowpea (Vigna unguiculata [Linn.] Walp) as determined by illumina MiSeq sequencing and culture-dependent methods. Current Microbiology, 78(4), 1286–1297. https://doi.org/10.1007/s00284-021-02382-3
- He, Z., Chen, H., Wang, X., Lin, X., Ji, C., Li, S., & Liang, H. (2020). Effects of different temperatures on bacterial diversity and volatile flavor compounds during the fermentation of suancai, a traditional fermented vegetable food from northeastern China. LWT, 118, 108773. https://doi.org/10.1016/j.lwt.2019.108773
- Huang, Z., Yu, X., Yang, Q., Zhao, Y., & Wu, W. (2021). Aptasensors for Staphylococcus aureus risk assessment in food. Frontiers in Microbiology, 12, 714265. https://doi.org/10.3389/fmicb.2021.714265
- Hu, N., Lei, M., Zhao, X., Wang, Y., Zhang, Y., & Wang, S. (2020). Analysis of microbiota in Hainan Yucha during fermentation by 16S rRNA gene high‐throughput sequencing. Journal of Food Processing and Preservation, 44(7), e14523. https://doi.org/10.1111/jfpp.14523
- Jiang, S., Ma, C., Peng, Q., Huo, D., Li, W., & Zhang, J. (2019). Microbial profile and genetic polymorphism of predominant species in some traditional fermented seafoods of the Hainan area in China. Frontiers in Microbiology, 10, 564. https://doi.org/10.3389/fmicb.2019.00564
- Jun, Z., Shuaishuai, W., Lihua, Z., Qilong, M., Xi, L., Mengyang, N., Tong, Z., & Hongli, Z. (2018). Culture-dependent and-independent analysis of bacterial community structure in Jiangshui, a traditional Chinese fermented vegetable food. LWT, 96, 244–250. https://doi.org/10.1016/j.lwt.2018.05.038
- Kim, S. S., Kwak, H. S., & Kim, M. J. (2020). The effect of various salinity levels on metabolomic profiles, antioxidant capacities and sensory attributes of doenjang, a fermented soybean paste. Food Chemistry, 328, 127176. https://doi.org/10.1016/j.foodchem.2020.127176
- Lee, M. A., Choi, Y. J., Lee, H., Hwang, S., Lee, H. J., Park, S. J., Chung, Y. B., Yun, Y. R., Park, S. H., & Min, S. (2021). Influence of salinity on the microbial community composition and metabolite profile in Kimchi. Fermentation, 7(4), 308. https://doi.org/10.3390/fermentation7040308
- Lemos, L. N., Fulthorpe, R. R., Triplett, E. W., & Roesch, L. F. (2011). Rethinking microbial diversity analysis in the high throughput sequencing era. Journal of Microbiological Methods, 86(1), 42–51. https://doi.org/10.1016/j.mimet.2011.03.014
- Liang, T., Xie, X., Zhang, J., Ding, Y., & Wu, Q. (2021). Bacterial community and composition of different traditional fermented dairy products in China, South Africa, and Sri Lanka by high-throughput sequencing of 16S rRNA genes. LWT, 144, 111209. https://doi.org/10.1016/j.lwt.2021.111209
- Li, Q., Liu, X., Dong, M., Zhou, J., & Wang, Y. (2015). Adhesion and probiotic properties of Lactobacillus plantarum isolated from Chinese traditional fermented soybean paste. Global Advanced Research Journal of Food Science and Technology, 4(1), 001–009.
- Lin, L., Wu, J., Chen, X., Huang, L., Zhang, X., & Gao, X. (2020). The role of the bacterial community in producing a peculiar smell in Chinese fermented sour soup. Microorganisms, 8(9), 1270. https://doi.org/10.3390/microorganisms8091270
- Liu, Z., Li, J., Wei, B., Huang, T., Xiao, Y., Peng, Z., Xie, M., & Xiong, T. (2019). Bacterial community and composition in Jiang-shui and Suan-cai revealed by high-throughput sequencing of 16S rRNA. International Journal of Food Microbiology, 306, 108271. https://doi.org/10.1016/j.ijfoodmicro.2019.108271
- Liu, Z., Peng, Z., Huang, T., Xiao, Y., Li, J., Xie, M., & Xiong, T. (2019). Comparison of bacterial diversity in traditionally homemade paocai and Chinese spicy cabbage. Food Microbiology, 83, 141–149. https://doi.org/10.1016/j.fm.2019.02.012
- Parvez, S., Malik, K. A., Ah Kang, S., & Kim, H. Y. (2006). Probiotics and their fermented food products are beneficial for health. Journal of Applied Microbiology, 100(6), 1171–1185. https://doi.org/10.1111/j.1365-2672.2006.02963.x
- Peng, Q., Jiang, S., Chen, J., Ma, C., Huo, D., Shao, Y., & Zhang, J. (2018). Unique microbial diversity and metabolic pathway features of fermented vegetables from Hainan, China. Frontiers in Microbiology, 9, 399. https://doi.org/10.3389/fmicb.2018.00399
- Poirier, S., Rué, O., Peguilhan, R., Coeuret, G., Zagorec, M., Champomier-Verges, M. C., Loux, V., & Chaillou, S. (2018). Deciphering intra-species bacterial diversity of meat and seafood spoilage microbiota using gyrB amplicon sequencing: A comparative analysis with 16S rDNA V3-V4 amplicon sequencing. Plos One, 13(9), e0204629. https://doi.org/10.1371/journal.pone.0204629
- Salvetti, E., Torriani, S., & Felis, G. E. (2012). The genus Lactobacillus: A taxonomic update. Probiotics and Antimicrobial Proteins, 4(4), 217–226. https://doi.org/10.1007/s12602-012-9117-8
- Song, N. E., Kim, N. J., Kim, Y. H., & Baik, S. H. (2021). Probiotic properties of lactic acid bacteria with high conjugated linoleic acid converting activity isolated from jeot-gal, high-salt fermented seafood. Microorganisms, 9(11), 2247. https://doi.org/10.3390/microorganisms9112247
- Teixeira, C. G., Fusieger, A., Milião, G. L., Martins, E., Drider, D., Nero, L. A., & de Carvalho, A. F. (2021). Weissella: An emerging bacterium with promising health benefits. Probiotics and Antimicrobial Proteins, 13(4), 915–925. https://doi.org/10.1007/s12602-021-09751-1
- Ward, T., Larson, J., Meulemans, J., Hillmann, B., Lynch, J., Sidiropoulos, D., Spear, J. R., Caporaso, G., Blekhman, R., & Knight, R. (2017). BugBase predicts organism-level microbiome phenotypes. bioRxiv, 133462.
- Xiong, T., Guan, Q., Song, S., Hao, M., & Xie, M. (2012). Dynamic changes of lactic acid bacteria flora during Chinese sauerkraut fermentation. Food Control, 26(1), 178–181. https://doi.org/10.1016/j.foodcont.2012.01.027
- Yan, Y., Chen, H., Sun, L., Zhang, W., Lu, X., Li, Z., Xu, J., & Ren, Q. (2022). The changes of microbial diversity and flavor compounds during the fermentation of millet Huangjiu, a traditional Chinese beverage. Plos One, 17(1), e0262353. https://doi.org/10.1371/journal.pone.0262353
- Yu, H. S., Jang, H. J., Lee, N. K., & Paik, H. D. (2019). Evaluation of the probiotic characteristics and prophylactic potential of Weissella cibaria strains isolated from kimchi. LWT, 112, 108229. https://doi.org/10.1016/j.lwt.2019.05.127
- Zhang, Y. Z., Lin, X. N., Ji, Y. Q., He, H. J., Yang, H. Z., Tang, X. J., & Liu, Y. G. (2022). Characterization and correlation of dominant bacteria and volatile compounds in post-fermentation process of Ba-bao Douchi. Food Research International, 160, 111688. https://doi.org/10.1016/j.foodres.2022.111688
- Zhang, J., Wang, X., Huo, D., Li, W., Hu, Q., Xu, C., Liu, S., & Li, C. (2016). Metagenomic approach reveals microbial diversity and predictive microbial metabolic pathways in Yucha, a traditional Li fermented food. Scientific Reports, 6(1), 1–9. https://doi.org/10.1038/srep32524
- Zhao, C., Su, W., Mu, Y., Jiang, L., & Mu, Y. (2020). Correlations between microbiota with physicochemical properties and volatile flavor components in black glutinous rice wine fermentation. Food Research International, 138, 109800. https://doi.org/10.1016/j.foodres.2020.109800
- Zhou, X., Liu, Z., Xie, L., Li, L., Zhou, W., & Zhao, L. (2022). The correlation mechanism between dominant bacteria and primary metabolites during fermentation of red sour soup. Foods, 11(3), 341. https://doi.org/10.3390/foods11030341