
ABSTRACT
The World Health Organization (WHO) has identified antimicrobial resistance (AMR) as one of the top three global dangers to public health. One of the most vital factors contributing to the high prevalence of AMR is the misuse/overuse of antibiotics for treatment and/or as a growth promoter in the food industry. AMR can be transmitted to humans via food, the environment, or other channels through horizontal gene transfer. Therefore, efficient methods are urgently needed to determine whether bacteria are resistant to antibiotics. This work provides a review of the advances in machine learning (ML) techniques for predicting and identifying AMR in foodborne pathogens. We also emphasize the groundbreaking potential of whole genome sequencing (WGS) and spectroscopy technologies combined with ML in the context of AMR detection. These offer enormous potential because of their unique characteristics, which can overcome inherent limits in existing detection approaches.
1. Introduction
Antibiotics are considered to have made significant contributions to medicine and humanity in the 20th century due to their effectiveness in killing or inhibiting the growth of bacteria (Uddin et al., Citation2021). For decades, antibiotics have been used not only for medical purposes, but also as a precaution in various fields including livestock and agriculture (Mann et al., Citation2021). However, the abuse of antibiotics in the food industry has resulted in the emergence of antimicrobial resistance (AMR), which can be spread to the public (Economou & Gousia, Citation2015). Food plays a significant role in continuous transmission of AMR (Stratev & Odeyemi, Citation2016). AMR among foodborne pathogens poses a serious concern to public health because it interferes with the action of antibiotics and increases the rate of ineffective and unsuccessful antibiotic therapy (Grudlewska-Buda et al., Citation2023). Therefore, it is crucial to determine whether a strain is resistant or sensitive to antibiotics to combat AMR foodborne pathogens. If they are not treated correctly, AMR foodborne pathogens impose a significant financial burden on society, leading to suffering, discomfort, and ultimately fatalities (Yasir et al., Citation2022).
In traditional methods, antimicrobial susceptibility testing (AST) primarily relies on culture-based methods, which are both time-consuming and labor-intensive (Anahtar et al., Citation2021). In contrast, machine learning (ML) can quickly identify AMR genes and enhance prediction accuracy using genomic-based and spectroscopy-based techniques. ML algorithms are a subfield of artificial intelligence that can develop the ability to accurately predict outcome variables from extensive input datasets (El Bouchefry & de Souza, Citation2020). Recently, the application of ML to address the issue of AMR has gained increased attention due to improved algorithm performance and the urgency of novel approaches to lowering the burden of foodborne diseases (Anahtar et al., Citation2021).
Therefore, in this review, we provided an overview of the emergence and transmission of AMR throughout the farm-to-table chain. We also showed that the rapid advancement of ML technology has provided opportunities for researchers to acquire information for intelligent data analysis. Particularly, ML in combination with whole genome sequencing (WGS) and spectroscopy technologies have been recently used for the identification of AMR. The reliability and accuracy of these high-throughput and affordable technologies allowed for the rapid prediction of AMR. They can be a powerful tool for fast screening the transmission of AMR genes, providing significant potential to alleviate the severity of AMR in the future.
2. AMR along the agri-food chain
2.1. Global trend in antibiotic use in the food industry
Antibiotic agents are widely used to treat infectious diseases in both humans and animals (McEwen & Fedorka-Cray, Citation2002). There are four main classifications for antibiotic usage in food animals: therapy, prophylaxis, metaphylaxis, and growth promotion (Bandyopadhyay & Samanta, Citation2020). These four categories can enhance the health of food animals and the agricultural environment, benefiting animal welfare (Economou & Gousia, Citation2015). However, there has been an unprecedented increase in the need for animal protein worldwide (Van Boeckel et al., Citation2015). The animal production systems have made it easier to meet this expanding demand by using antibiotics to maintain animal health and productivity (Hosain et al., Citation2021). This worldwide consumption of veterinary antibiotics was estimated at 99,502 tones in 2020, and it is expected to rise by 8.0% to 107,472 tones by 2030 (Mulchandani et al., Citation2023). The growing global use of antibiotics in animal protein drives AMR infections, which have serious health effects on both people and animals (Van Boeckel et al., Citation2017). According to the 2019 Interagency Coordination Group on Antimicrobial Resistance (IACG) report (WHO, Citation2019b), at least 700,000 deaths worldwide were already attributed to AMR each year and this number might rise to 10 million fatalities annually worldwide by 2050. The severity of AMR has prompted international organizations all around the world to try developing alternative tactics. In 2017, The World Health Organization (WHO) recognized the health risks associated with the improper use of veterinary antibiotics and strongly advised against using any kind of veterinary antibiotics, including those used for growth promotion and disease prevention without a diagnosis (WHO, Citation2017a). Further, the UN, international agencies, and experts issued a new report calling for swift, concerted, and aggressive action to prevent a catastrophic drug-resistance disaster in 2019 (WHO, Citation2019a). This report recommends a coordinated, multisectoral “One Health” approach, emphasizing the tight connections between the health of people, animals, food, plants, and the environment.
2.2. AMR in common foodborne pathogens
AMR of foodborne bacteria and its reservoirs in food animals, such as chickens, pigs, and cattle have emerged as major worldwide public health issues (Koch et al., Citation2017). The WHO produced a list of antibiotic-resistant “priority pathogens” in 2017 (WHO, Citation2017b). Salmonella spp., Campylobacter spp., and Staphylococcus aureus were listed as the high-priority groups that provide the greatest risk to human health due to their AMR (WHO, Citation2017b). This section aims to provide insights into current knowledge of the AMR foodborne pathogens mentioned above and their prevalence along the food chain.
2.2.1. Salmonella spp
Salmonella spp. is a foodborne pathogen transmitted to humans through the consumption of contaminated poultry, eggs, and dairy products (Ehuwa et al., Citation2021). Recently, an increase in AMR Salmonella along the food chain has posed a public health concern worldwide. For instance, the annual incidence of AMR in Salmonella spp. obtained from people, retail meat, and food animals in the United States has increased by an estimated 40% (Medalla et al., Citation2021). In particular, the number of Salmonella infections that are resistant to ciprofloxacin, ceftriaxone, or ampicillin rose from around 159,000 infections in 2004–2008 to roughly 222,000 infections in 2015–2016 (Medalla et al., Citation2021).
Additionally, the incidence of multidrug-resistant (MDR) Salmonella spp. is rising globally, and farm animals frequently act as reservoirs for MDR outbreaks (Doyle, Citation2015). Rodrigues et al. (Citation2020) identified 65 different resistance genes from 930 WGS of Salmonella based on the National Biotechnology Information Center’s (NCBI) public database. The most common ones were tet(A), sul2, and fosA7. This study stated that 58.0% (540/930) of the bacteria might present a MDR profile, underscoring the significance of MDR Salmonella. It is a serious problem that has also been observed in developing countries. Ramtahal et al. (Citation2022) showed that there is an apparent widespread usage of antibiotics and transmission of MDR Salmonella isolates are a major problem in Africa. MDR Salmonella isolates were discovered in 30 of the 64 studies reporting AMR, with prevalence ranging from 12.1% in Zimbabwe to 100% in Egypt, Ethiopia, Nigeria, Senegal, and South Africa. Among the 14 antibiotic classes for which resistant isolates were found, aminoglycosides, quinolones, penicillin, sulfonamides, diaminopyrimidines, and tetracyclines had the highest rate of resistance in this study.
2.2.2. Campylobacter spp
Campylobacter spp. is a major cause of AMR-related foodborne disease through the consumption of raw or undercooked meat, unpasteurized dairy products, and contaminated drinking water (Samtiya et al., Citation2022). The use of antibiotic drugs, particularly fluoroquinolones, in animal production is one of the primary factors causing AMR in Campylobacter spp. strains (Wieczorek & Osek, Citation2013). Campylobacter‘s major mechanism of fluoroquinolone resistance is a mutation in the Quinolone Resistance Determinant Region (QRDR) of the gyrA gene, which codes for a subunit of the enzyme DNA gyrase, which fluoroquinolone targets (Frasao et al., Citation2015). Espinoza et al. (Citation2020) also discovered that rising fluoroquinolone resistance rates in Campylobacter jejuni posed a significant public health risk. They discovered point mutations at T86I in the gyrA gene in all 141 clinical isolates of C. jejuni that had previously been determined as ciprofloxacin-resistant using the E-test.
Because of concerns about a rise in fluoroquinolone-resistant Campylobacter spp., fluoroquinolone usage restrictions for animal husbandry have been implemented in the EU (2006) and the U.S.A. (2005) (Nelson et al., Citation2007; Veltcheva et al., Citation2022). Nevertheless, the incidence of fluoroquinolone-resistant Campylobacter spp. is still rising quickly (Khademi et al., Citation2020). According to the 2019–2020 EFSA report, ciprofloxacin and nalidixic acid were shown to have the highest levels of resistance in broiler meat, with total percentages of 64.2–90.0% in 2019 and 71–84.8% in 2020 in the EU Member States (MSs) (EFSA & ECDC, Citation2022). The resistance of Campylobacter jejuni and Campylobacter coli to ciprofloxacin from broilers has increased significantly in twelve MSs and two non-MSs, whereas there has only been a drop in Spain, which has become a major public health concern in the European Union. Since ciprofloxacin is one of the critically important antimicrobials (CIA) used to treat human Campylobacter spp. infections, ciprofloxacin-resistant C. jejuni and C. coli from both humans and animals are particularly relevant for public health (EFSA & ECDC, Citation2022).
2.2.3. Staphylococcus aureus
Staphylococcus aureus is a common Gram-positive bacterium found on the skin and mucosa of both humans and animals (Haag et al., Citation2019). With the discovery of penicillin in the 1940s, S. aureus was one of the initial bacteria to be identified as being resistant to penicillin (Livermore, Citation2000). Around 1960, the MDR S. aureus was developed, along with the emergence of methicillin-resistant S. aureus (MRSA) (Turner et al., Citation2019). MRSA is the strain of S. aureus that is resistant to methicillin and other β-lactam antibiotics; they feature a new gene (mecA) that codes for the penicillin-binding protein 2a (PBP2a), a somewhat different PBP that has low affinity for β-lactam drugs (Larsen et al., Citation2022; Wang et al., Citation2017). Aside from mecA, the most prevalent AMR gene found was ermC, followed by ermB, blaZ, aacA-aphD, and ermA in MRSA isolates from food animals (Gan et al., Citation2021).
Before the 1990s, MRSA was initially found in a hospital environment, but it has become widely distributed among food-producing animals (Aires de Sousa, Citation2017; De Boer et al., Citation2009). In particular, livestock-associated methicillin-resistant S. aureus (LA-MRSA) is growing concern worldwide because people who have frequent, prolonged contact with livestock are more likely to contact LA-MRSA (Crespo-Piazuelo & Lawlor, Citation2021; Khademi et al., Citation2020). The incidence of LA-MRSA among livestock workers varies throughout the world, with reported rates for pig employees in Europe ranging from 24% to 86%, North America from 20% to 45%, and Asia from 6% to 19% (Anjum et al., Citation2019; C. Chen & Wu, Citation2021; Chuang & Huang, Citation2015; Golding et al., Citation2010; Khanna et al., Citation2008; Smith et al., Citation2009).
2.3. AMR dissemination throughout the agri-food chain
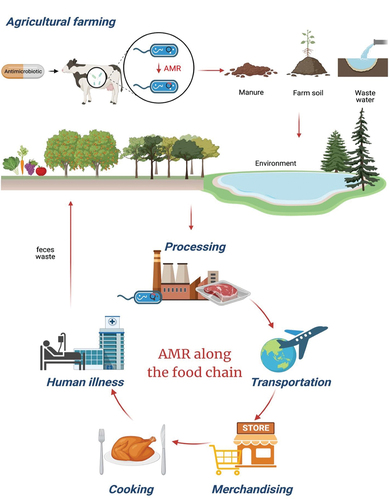
Recently, concerns regarding AMR have been exacerbated due to its transmission through the agri-food chain, including agricultural farming, processing, transportation, merchandising, and cooking (). Domesticated livestock and animal products containing AMR genes serve as the primary sources of AMR dissemination within the agri-food chain (Samtiya et al., Citation2022). AMR foodborne pathogens can spread through environmental pathways such as manure, farm soil, and wastewater, which act as “hot spots” for AMR pollution (Vinayamohan et al., Citation2022). Particularly, feces waste from animals and humans serves as a transmission route for AMR, as it can contaminate crops and contribute to runoff entering water systems (Vinayamohan et al., Citation2022). Additionally, there are several pathways through which food can get contaminated with AMR genes. Food handlers are associated with the spread of AMR, often due to inadequate hygiene practices or cross-contamination when handling food (Samtiya et al., Citation2022). Such cross-contamination may arise from improperly cleaned machinery and equipment, as well as from clogged, outdated ventilation filters during food processing (Freeland et al., Citation2023). Also, transportation has contributed to the increased risk of AMR cross-contamination. The global spread of AMR within the agri-food chain is facilitated by factors such as the expanding human population and the globalization of animals and food product trade (Samtiya et al., Citation2022). Finally, when food is handled by sellers and consumer, it can be contaminated with AMR bacteria. Trays, tables, and kitchen counters that have been improperly handled or inadequately packaged can potentially expose people to contaminated food and environments (Freeland et al., Citation2023)
Figure 1. Illustration of the potential routes of AMR in the food chain.
3. Mechanisms of bacterial AMR
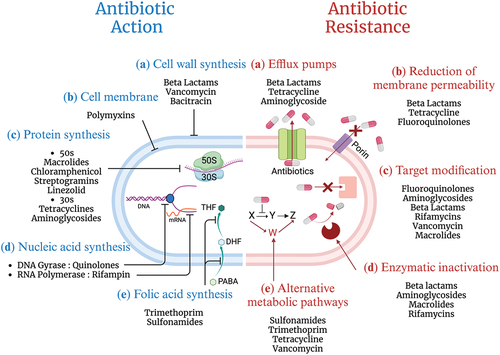
The misuse/abuse of antibiotics has led to an increase in the number of AMR bacteria, which have greatly endangered public health. In this section, the mechanisms underlying AMR are introduced to better understand how bacteria become resistant to antibiotics ().
Figure 2. The mechanism of antibiotic action (left). Antibiotics can inhibit the growth of bacteria by interfering with (1) the bacterial cell wall or (2) the cell membrane synthesis. Antibiotics also target (3) protein synthesis and (4) nucleic acid. (5) lastly, antibiotics can also function as antimetabolites by inhibiting the folate metabolism via a pathway involving para-aminobenzoic acid (PABA) and two folic acid precursors, dihydrofolate (DHF) and tetrahydrofolate (THF). Sulfonamide and trimethoprim are structural analogs of PABA and DHF that block dihydropteroate synthetase and dihydrofolate reductase, respectively, preventing bacteria from synthesizing folic acid, a component required for the synthesis of nucleotides. On the other hand, there are five categories of AMR mechanisms (right). (1) the efflux pumps export antibiotics from the cells to lower the concentration of antibiotics inside bacterial cells. (2) the permeability of the outer membrane can be reduced by porins that allow antibiotics to enter the bacterial cell passively. (3) target modification includes changing the antibiotic targets to lessen antibiotic binding. (4) antibiotics are rendered inactive by enzymes that either break down or alter the antibiotic molecule. (5) target bypass replaces the antibiotics target with a replacement protein that performs the same function without being inhibited by the antibiotics, rendering the antibiotics useless and the original target unnecessary.
3.1. Reduction of the inner and outer membrane permeability
Changes in the permeability of the bacterial outer membrane, which serves as a barrier between the bacteria and the outside environment, can result in AMR (Delcour, Citation2009). Particularly, this resistance mechanism is primarily found in Gram-negative bacteria, mainly due to changes in porins (Breijyeh et al., Citation2020). Porins are non-specific channels located in the outer membrane of Gram-negative bacteria, responsible for allowing the diffusion of antibiotics into the bacterial cells (Breijyeh et al., Citation2020). Small hydrophilic antibiotics such as β-lactams and quinolones can only pass the outer membrane of bacteria through porins (Delcour, Citation2009). Therefore, a reduction in outer membrane permeability consequently contributed to the evolution of AMR as β-lactams and quinolones cannot enter the cell.
3.2. Activation of efflux pumps
Efflux pumps are membrane proteins that actively expel antibiotic substances from the inside of the bacterial cell to the external environment (D. Du et al., Citation2018). AMR-related efflux pumps can be broadly divided into five classes: the major facilitator (MF) family, the adenosine triphosphate-binding cassette (ABC) family, the resistance-nodulation-division (RND) family, the small multidrug resistance (SMR) family, and the multidrug and toxic compound extrusion (MATE) family (Annunziato, Citation2019). Increased active efflux pumps are a serious problem since a single species of MDR efflux pump can transport a wide range of antibiotic agents out of the bacterial cell, facilitating the development of MDR bacteria (Nishino et al., Citation2021).
3.3. Enzymatic inactivation of the antibiotics
Enzymatic inactivation of the antibiotics is one of the most significant mechanisms of resistance and there are two main ways of this mechanism; (1) actual degradation of the antibiotics and (2) transfer of a chemical group to the antibiotics (Reygaert, Citation2018). In the former, the β-lactamase enzyme is one of the most prevalent hydrolytic enzymes. It hydrolyzes the β-lactam ring of β-lactam antibiotics such the cephalosporins, which are mostly of concern in Gram-negative bacteria (Bonomo, Citation2017; Bush, Citation2010). While the most widely used methods for transferring chemical groups are acetylation, phosphorylation, and adenylation (Reygaert, Citation2018). In this mechanism, the enzymes chemically alter the antibiotics by adding a chemical group to them, rendering the drug ineffective. The most often employed method is acetylation, which is known to be effective in combating streptogramins, chloramphenicol, and aminoglycosides. Phosphorylation is often employed for aminoglycosides and chloramphenicol, while adenylation is used to address lincosamides and aminoglycosides (Munita & Arias, Citation2016).
3.4. The modifications of the antibiotic binding targets
Most antibiotics have high-affinity particular bindings to their targets, inhibiting the target’s usual activities (Dhanda et al., Citation2023). AMR can emerge as a result of target structural modifications that hinder effective antibiotic binding (Giedraitienė et al., Citation2011). In this mechanism, the resistance to the β-lactams serves as a prominent example. The antibacterial action of β-lactams depends on their capacity to prevent the formation of cell walls by inhibiting penicillin-binding proteins (PBPs), which are significant enzymes in charge of the formation of cross-links between peptidoglycan units (Uddin et al., Citation2021). Modifications in the structure of PBPs can lead to resistance to β-lactam by weakening the attraction between the antibiotic agents and the PBPs (Sun et al., Citation2014).
3.5. Using an alternative metabolic pathway
Another mechanism of AMR involves circumventing the necessity for a specific target. Folic acid is an essential component for the metabolism of nucleic acids (DNA and RNA) and amino acids (Blancquaert et al., Citation2010). antibiotics like trimethoprim and sulfonamide interfere with the synthesis of nucleic acids and amino acids by mimicking essential substrates in the folic acid metabolism (Kapoor et al., Citation2017). However, bacteria can adapt by utilizing alternative metabolic pathways instead of folic acid synthesis to develop resistance to sulfonamide and trimethoprim (Urban-Chmiel et al., Citation2022).
4. The fundamentals of ML for AMR prediction
4.1. Sequencing data for ML
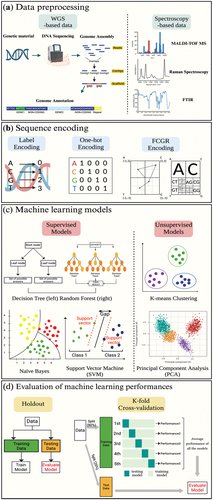
Genotypic ML models typically require high-quality sequencing data in which shotgun DNA sequences are commonly used (Anahtar et al., Citation2021). Therefore, it is crucial to organize the sequencing dataset through genome assembly and annotation as preliminary steps for predicting AMR (Boolchandani et al., Citation2019). DNA sequencing can be converted into numbers by encoding because ML algorithms can only identify numeric variables as input (Yang et al., Citation2020). Then, researchers can select the appropriate ML models to understand patterns and relations of the data that can make accurate predictions or classifications. Lastly, the holdout and k-fold cross-validation can assess the performance of ML models on a dataset (Xiong et al., Citation2020).
4.1.1. Genome assembly
There are various types of sequencing platforms. Illumina DNA sequencing produces short reads that accurate but result in fragmented genome assemblies (Wick, Judd, Gorrie, Holt, & Phillippy, Citation2017). Long reads of the DNA sequencing from Pacific Biosciences and Oxford Nanopore Technologies provide complete genome assemblies, but they are error-prone (Wick, Judd, Gorrie, Holt, & Phillippy, Citation2017). DNA sequencing can be represented as text sequences (reads), which are composed of four kinds of nucleotide initials A, C, G, and T (Chin et al., Citation2014). Therefore, it is crucial to reconstruct reads into longer and contiguous sequences (contigs) to produce the complete sequence. This procedure, known as genome assembly, is one of the subsequent phases in DNA sequencing () (Q. Chen et al., Citation2017). In recent times, hybrid assemblies, which combine both short and long reads, have gained popularity as a way to compensate for their shortcomings (Wick, Judd, Gorrie, Holt, & Phillippy, Citation2017). The Unicycler, a new tool designed for hybrid assemblies, uses SPAdes to produce the initial assembly from short reads. Subsequently, a simplified graph is created by integrating data from short and long reads together (Wick, Judd, Gorrie, & Holt, Citation2017; Wick, Judd, Gorrie, Holt, & Phillippy, Citation2017). As a result, hybrid assemblies offer a comprehensive dataset, encompassing crucial information on AMR and mobility genes that are pivotal for addressing AMR (Khezri et al., Citation2021).
Figure 3. Overview of ML for AMR prediction (a) genome assembly is the process of rebuilding the whole DNA sequence from the short, fragmented sequences. Genome annotation is the act of identifying and elucidating the functionalities of the bacteria. On the other hand, the MALDI-TOF MS, Raman spectroscopy, and FTIR are utilized for preprocessing in the context of spectroscopy-based ML applications. (b) Genome encoding refers to the process of delivering genetic data uniformly so that computers can understand and evaluate it. (c) ML models find underlying patterns of datasets for predicting new, unseen data. (d) Both holdout validation and k-fold cross-validation are crucial strategies for figuring out how well a ML model will perform on new, unseen data.
4.1.2. Genome annotation
The final genome assemblies need to be annotated after evaluation with QUAST (Quality Assessment Tool) (Gurevich et al., Citation2013). Genome annotation is the process of identifying biological information, such as the sequence of AMR genes in bacteria () (Gupta et al., Citation2014; Stein, Citation2001). This procedure converts raw genomic data into a format that can be used for further analysis and provides comprehensible information on the biological functions (). Recently, information systems have rapidly developed for the quick annotation of WGS. As an example, the BV-BRC offers researchers a private workspace for bioinformatics services such as genome assembly and annotation through the integration of a wealth of pathogen data with analytic tools () (Davis et al., Citation2020). The Genome Annotation Service in the BV-BRC makes use of a variety of tools, including the RAST tool kit (RASTtk), Viral Genome ORF Reader (VIGOR4), and PHANOTATE to annotate genomic characteristics in bacteria, viruses, and bacteriophages, respectively (Olson et al., Citation2023).
Figure 4. Information available for genomes with the Genome Annotation Service in BV-BRC. (a) The genome landing page. (b) Genome browser. (c) Specialty gene page. (d) Phylogeny.. (e) Circular view of the genome. Researchers can search for genes in their genome that are related to AMR, virulence factor.
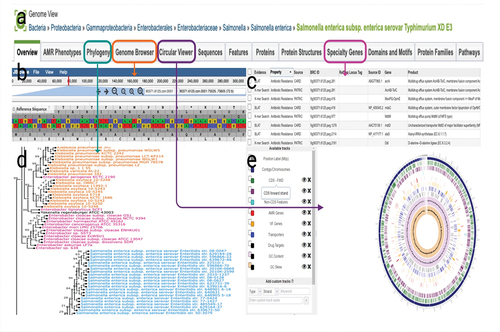
Figure 5. The bacterial and viral bioinformatics resource center (BV-BRC) (https://www.bv-brc.org/). (a) BV-BRC’s genome assembly landing page where researchers can assemble either single- or paired-end short reads, or longer reads. On the right side with an enlarged figure, there are listings of the assembly tools available. (b) The genome annotation landing page where researchers can annotate assemblies.
4.2. Sequence encoding
ML algorithms require numerical variables as input datasets (Spänig & Heider, Citation2019). Therefore, it is crucial to encode DNA sequencing using methods like label encoding, one-hot encoding, and Frequency Chaos Game Representation (FCGR) to create numeric input variables (Y. Ren et al., Citation2022) (). Genetic information in DNA is represented by long sequences of nucleotides with the four letters A, T, C, and G. Through label encoding, these DNA sequences can be transformed into an appropriate input variables by converting categorical formats into numeric variables (Y. Ren et al., Citation2022). One-hot coding and FCGR are commonly used for DNA classification through sequence mapping. In one-hot coding, DNA nucleotides are mapped to binary numbers, such as A = (1 0 0 0), G = (0 1 0 0), C = (0 0 1 0), and T = (0 0 0 1) (Q. Zhang et al., Citation2019). FCGR is another method of DNA sequencing mapping that maintains sequence patterns (Abd-Alhalem et al., Citation2020). This encoding calculates the distance between genomes to produce phylogenetic trees (Hatje & Kollmar, Citation2012).
4.3. ML models
Supervised learning, unsupervised learning, and reinforcement learning are the three categories of ML models. Supervised learning uses a set of input factors to predict a corresponding target output, known as a label. Unsupervised learning uses unlabeled data, so there are no corresponding output labels. Reinforcement learning, on the other hand, focuses primarily on rewards and penalties (Anahtar et al., Citation2021). In brief, the agent learns by being rewarded for positive acts and punished for negative ones. As a result, its usefulness in predicting AMR is limited. This section offers a comprehensive understanding of supervised learning and unsupervised learning that is widely used for the identification of AMR ().
4.3.1. Supervised learning models
Supervised learning models are trained using labeled datasets to find the best-fitting model for predicting outcomes with high precision, accuracy, or recall (Schmitt et al., Citation2020). One of the supervised learning algorithms is the decision tree (DT). In a DT, a training dataset is divided into branches that are then subdivided until a leaf node is reached (Song & Ying, Citation2015). A random forest (RF) comprises multiple randomly generated DTs, resulting in more precise predictions as the number of trees increases (Adnan & Islam, Citation2016; Breiman, Citation2001). Additionally, the RF algorithm trains its “forest” through bagging, an ensemble meta-algorithm that increases the random forest’s accuracy (Breiman, Citation2001). AdaBoost (Adaptive Boosting) and XGBoost (Extreme Gradient Boosting) are two well-known ensemble techniques. AdaBoost classifier begins by fitting a classifier on the initial data set, and then starts training a duplicate of the same classifier on an updated dataset. Updated data sets can concentrate on more incorrect classifiers to balance incorrect and error-prone data pieces (Azmi & Baliga, Citation2020). The XGBoost Classifier model is a simplified group based on the Gradient Boosting Decision Tree (GBDT) (Azmi & Baliga, Citation2020). It is an ensemble approach in which new models are built to correct the residuals or mistakes of previous models, which are then integrated to form the final prediction (Asselman et al., Citation2023). Another well-known supervised learning model is the support vector machine (SVM), which functions by creating margins between classes (Chauhan et al., Citation2019). The margins are designed to minimize the classification error by increasing the distance between the margin and the classes (Awad et al., Citation2015). SVM-RBF (Radial basis function) is an extension of SVM that uses the RBF kernel (Yuan et al., Citation2023). The RBF kernel enables SVM-RBF to handle non-linearly separable data by transferring the input features into a higher-dimensional space, allowing SVM-RBF to capture complex relationships and nonlinear decision boundaries. Lastly, Naïve Bayes (NB) is a classification technique with an assumption that the features of the input data are conditionally independent in a class, allowing the algorithm to predict outcomes rapidly and precisely (Al-Aidaroos et al., Citation2010). The classifier can perform effectively in challenging situations when the data distribution is not well-defined, as the NB technique entails a kernel function to calculate the probability density function of the input data (Ren et al., Citation2009).
4.3.2. Unsupervised learning models
Unsupervised learning makes use of unlabeled data as a training dataset to identify patterns and trends in raw datasets. Unsupervised learning is an effective method for understanding relationships in datasets by grouping data along similar features (Sarker, Citation2021). K-means is one of the most straightforward unsupervised learning techniques (Sinaga & Yang, Citation2020). The input datasets are divided into k clusters, and one is randomly selected from the input datasets. In the following step, algorithms determine the separations between each data point and the cluster centroid (Sarker, Citation2021). These distances are then calculated using algorithms to classify a set of data into a specific number of clusters. The principal component analysis (PCA) is another unsupervised machine learning technique for facilitating calculations. By lowering the dimensionality of the data, this approach makes it possible to observe variation from two-dimensional or three-dimensional data (T. Y. Du, Citation2019). It is used to provide a linear explanation for the variance-covariance structure of a set of variables (Z. Zhang & Castelló, Citation2017).
4.4. Evaluation of ML performances
Model performance evaluation is necessary to evaluate model performance on unseen data and choose an unbiased model. The holdout and k-fold cross-validation are the commonly used methods for assessing the performance of ML models () (Xiong et al., Citation2020). The holdout separates the entire dataset into training and testing sets (Kourou et al., Citation2015). The training sets are used to train the model while the testing sets are used to evaluate the performance of the model on unseen examples of data. However, the holdout method is sensitive to the way datasets are split. The model is easily biased so that the separated training and validation sets can lead to inconsistent performance (Yadav & Shukla, Citation2016). The k-fold cross-validation is an alternative to holdout in another way of splitting the data. The data set is divided into k folds at random using this procedure (Yadav & Shukla, Citation2016). Then, k-1 folds are used for training models and the remaining fold is used for testing models. It is possible to obtain k different models by repeating k times and calculating the average performance of all the models. Thus, k-fold cross-validation reduces bias by offering a better assessment of the model performance.
5. The applications of ML for the identification of AMR in foodborne pathogens
The spread of AMR via the food supply chain represents a serious threat to public health (Hudson et al., Citation2017). It could result in global threats to human health, water, soil, as well as food safety (UN, Citation2020). Traditional methods such as disk-diffusion, E-test, and broth microdilution are regarded as the gold standards for determining AMR, but these methods require a lot of time and effort (Salam et al., Citation2023). Therefore, the development of novel methods for rapidly assessing the AMR of these foodborne bacteria is of utmost importance. Recently, ML in conjunction with WGS and spectroscopy-based approaches has opened new possibilities for the quick and accurate detection of AMR. This section aims to provide a wide variety of ML applications using the technologies to discover AMR of foodborne bacteria.
5.1. ML applications of WGS-based analysis
Unlike conventional AST, WGS can give information on bacterial characteristics in terms of resistance genes, plasmids, and virulence factor (Bonvegna et al., Citation2022). Such WGS data are essential for tracking and monitoring AMR because they can quickly and accurately improve the understanding of the genetic basis for dissemination pathways. Additionally, combining specific software tools with ML holds promise for the identification and prediction of AMR, harnessing the capability of ML to utilize vast databases containing hundreds or thousands of pathogen genomes ().
Table.1. Applications of machine learning (ML) using whole genome sequencing (WGS)-based data for AMR prediction.
5.1.1. WGS for the resistance profiles
The advancement of WGS technology has made it feasible to obtain genes for predicting the resistance phenotype (Rokney et al., Citation2020). Recently, AMR prediction has become attainable through the utilization of three widely recognized web tools: AMRFinderPlus, ResFinder, and the Comprehensive Antibiotic Resistance Database (CARD). The National Center for Biotechnology Information (NCBI) developed a tool called AMRFinder to find AMR genes in bacterial genomes (Feldgarden et al., Citation2019). Recently, NCBI launched AMRFinderPlus, a new version of AMRFinder that uses assembled and/or annotated nucleotide sequences to find AMR genes, resistance-associated point mutations, and certain classes of genes. Another tool, ResFinder is widely used for detecting horizontally acquired resistance genes in WGS data (Yadav & Kapley, Citation2021). Under the ResFinder 4.0 project, PointFinder was incorporated into ResFinder to identify mutations that confer resistance, providing a more comprehensive database of AMR determinants (Papp & Solymosi, Citation2022). These tools have begun to integrate ML-based AMR prediction to improve tool accuracy and extend the availability of the number of species and antibiotic combinations (Florensa et al., Citation2022). The CARD is a bioinformatic database that provides reference DNA and protein data on AMR-related genes (Alcock et al., Citation2020). The CARD database is updated regularly by skilled curators who evaluate the scientific literature. Their work is supplemented by a ML algorithm (CARD*Shark) that arranges scientific papers by reference for the process (Papp & Solymosi, Citation2022). Additionally, the Resistance Gene Identifier (RGI) utilizes the reference data from CARD to predict AMR genes from genome sequencing (McArthur et al., Citation2013). The Antibiotic Resistance Ontology (ARO), which serves as the central ontology within CARD, can be integrated with the RGI. ARO provides comprehensive explanations of AMR (Jia et al., Citation2017).
Maguire et al. (Citation2019) employed a logistic regression (LR) model to predict the AMR phenotype and identify the primary AMR drivers of Salmonella enterica isolated from broiler chickens. A total of 97 genomes were assembled, and RGI was then utilized to identify the AMR genes. The RGI predictions were contrasted with the AST results to compare phenotype to genotype. A simple binary LR model was built using the RGI-detected AMR determinants as the input features and “susceptible” and “resistant” as the output labels. Standard FDA criteria were then applied to determine the efficiency of the genotype-based ML technique in predicting AST. These findings were classified into three categories: categorical agreement (both the genomic data and the AST predicted susceptibility or resistance), major disagreement (the genomic data predicted resistance, but the AST indicated susceptibility), and very major disagreement (the genomic data predicted susceptibility, but the AST revealed resistance). As a result, they were able to predict the AMR phenotype with high accuracy (0.92 to 0.99) for 7 antibiotic agents with 80% of the training data. Additionally, they determined the primary drivers of AMR in these isolates by integrating the inferred k-mers and LR weights. They demonstrated that the AmpC-like CMY-2-β-lactamase was a principal driver of β-lactam resistance and that the phosphotransferase APH(6)-Id and APH(3″-Ib) were major drivers of streptomycin resistance in broiler chicken-isolated nontyphoidal S. enterica serovars.
5.1.2. WGS for the separation of plasmid sequence
It has difficulty identifying plasmid contigs in WGS data because it is challenging to distinguish whether it is from a plasmid or chromosome in a fragmented WGS (Tang et al., Citation2023). Therefore, it is crucial to determine whether the AMR gene is located on a more stable chromosome or a more mobile plasmid for monitoring and observing AMR (Andreopoulos et al., Citation2022). Some tools have been used to separate plasmid sequences from chromosomal contigs using ML. As an example, Plasmer is a reliable and accurate technique for identifying plasmid contigs in bacterial genome assemblies (Zhu et al., Citation2023). Plasmer effectively separates plasmid and chromosomal contigs by combining the benefits of shared k-mers and genomic characteristics, such as alignment E value and replicon distribution scores (RDS). Another tool called the mlplasmids (Arredondo-Alonso et al., Citation2018) is used to identify and extract plasmid sequences from WGS data. Also, it can serve as the foundation for the categorization of plasmids by other tools like PlacnetW, which facilitates plasmid sequence reconstruction.
Arredondo-Alonso et al. (Citation2018) introduced mlplasmids, a technique for determining if a certain contig is derived from a plasmid or chromosome. Initially, they downloaded complete genome sequences of Enterococcus faecium, Escherichia coli, and Klebsiella pneumoniae from the NCBI database. Using bwa-mem, SPAdes contigs were mapped against complete chromosomal and plasmid sequences to label them as either chromosome- or plasmid-derived. The SPAdes-labeled contigs for each bacterial species were split into training (80%) and test sets (20%) to train the five distinct supervised methods: Bayesian classifiers, LR, DT, RF, and SVM with 10-fold cross-validation. Consequently, SVM was the best classifier for predicting plasmid-derived contigs in the three bacterial species and they integrated the SVM models for E. faecium, K. pneumoniae, and E. coli in a new R package named “mlplasmids.”
5.1.3. WGS for the identification of virulence factors
When bacteria are found in an antibiotic environment, some bacteria developed their virulence which allows them to cause a variety of diseases (Beceiro et al., Citation2013). It is crucial to identify the virulence factors (VFs) because different foodborne pathogens with virulence genes generate a variety of disease endpoints, including mortality (Bintsis, Citation2017). ML deals with the increasing mass of bacterial genome sequence data and provides an opportunity for predicting and identifying bacterial VFs from genetic diversity (Uelze et al., Citation2020). Recently, the DeepVF which is a user-friendly online tool has been developed for screening and identifying VFs from genome sequence data (Xie et al., Citation2021). Unlike other tools which require the protein sequences in FASTA, the DeepVF is available with raw sequences as well, so that it can be automatically standardized into FASTA format. Additionally, the improved accuracy of DeepVF predictors can deal with the difficulties of identifying virulence determinants when they continually evolve with the AMR.
Xie et al. (Citation2021) exhibited the utility of DeepVF to achieve accurate identification of VFs. They collected 9749 VFs from three public databases such as Victors, VDFB, and Pathosystems Resource Integration Center (PATRIC) which offer comprehensive data sources for VF prediction. In the positive and negative datasets, the sequences were clustered and sequence redundancy was eliminated using the CD-HIT tool, respectively. Consequently, 3576 VFs and 4910 non-VFs made up the final non-redundant dataset. Of these, 3000 VFs and 4334 non-VFs were used as the training dataset (86%), while 576 VFs and 576 non-VFs were randomly selected to serve as the independent test dataset (14%). These datasets were utilized to investigate four classical ML algorithms: RF, SVM, XGBoost, and multilayer perceptron (MLP), as well as three deep learning algorithms: convolutional neural networks (CNNs), long short-term memory networks (LSTMs), and deep neural networks (DNNs). Subsequently, they established 62 baseline models, comprising 30 (3 × 10) Deep learning baseline models and 32 (4 × 8) ML baseline models and constructed a meta-model with XGBoost.
5.2. ML applications of spectroscopy techniques-based analysis
Applications of spectroscopy methods including matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS), Raman spectroscopy (RS), and Fourier-transform infrared spectroscopy (FTIR) have grown in popularity recently (Dieckmann et al., Citation2016). As a substitute for a traditional AMR detection technique, spectroscopy technologies allow for the quicker, easier, and more affordable AMR analysis. Additionally, by combining spectroscopy methods with ML, bigger datasets for a variety of microbial species can be provided to establish the AMR profiles. This section gives an overview of the progress and applications of spectroscopy methods in combination with ML for the identification of AMR.
5.2.1. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS)
One of the most used spectrometry tools in biology is the MALDI-TOF MS, which swiftly and precisely identifies a variety of bacterial genera and species (Hou et al., Citation2019). Additionally, the MALDI-TOF MS is a powerful tool that provides a rapid and precise way of detecting and evaluating AMR (Feucherolles et al., Citation2022). In order to identify AMR, a variety of approaches are being developed, including those for detecting labeled amino acids, antibiotics degradation, and AMR strain biomarkers (Hou et al., Citation2019).
Due to the ease of analyzing larger datasets, previous studies demonstrated that MALDI-TOF MS combined with ML can be a useful method for quickly and accurately screening AMR of foodborne pathogens (Feucherolles et al., Citation2022). Esener et al. (Citation2021) determined resistance to either multidrug or benzylpenicillin in Staphylococcus aureus isolates using MALDI-TOF MS and ML. They examined a collection of 82 S. aureus isolates obtained from 67 cows that had been diagnosed with bovine mastitis. The AMR profiles of the S. aureus isolates were categorized as either susceptible or resistant based on their minimum inhibitory concentration (MIC) to 18 antibiotics. The examination of MDR was carried out using 15 MDR isolates and 51 susceptible isolates, whereas the spectra of the 16 benzylpenicillin-resistant isolates and the 51 susceptible isolates were first pre-processed for analysis of benzylpenicillin. Utilizing MATLAB Bioinformatics Toolbox Release 2017b for pre-processing, four and five distinct peaks were discovered to appear in at least 30% of the total number of spectra for multidrug and benzylpenicillin, respectively. Then, 10 different supervised machine learning algorithms were constructed using these peaks as features to determine whether the isolates’ MALDI-TOF peaks could be utilized to predict the susceptibility or resistance to multidrug and benzylpenicillin. Prior to classification, isolates that were resistant and susceptible were labeled as 0 and 1, respectively, and each classifier was trained using 30 iterations of nested cross-validation. The multidrug and benzylpenicillin individual isolates were used to train the LDA and RBF SVM algorithms, which produced the highest accuracy rates of 96.81%±0.43% and 97.54 ± 1.91%, respectively. Also, Feucherolles et al. (Citation2022) assessed the possibility of ML in conjunction with MALDI-TOF MS to identify AMR in foodborne bacteria. In this study, seven different antibiotics were used to evaluate the AST of 224 Campylobacter jejuni and 116 Campylobacter coli that were acquired from humans, surface water, and animals (raccoons, wild birds, cattle, pigs, and poultry). Through mass spectra analysis of MALDI-TOF MS, peak matching was performed on Main spectrum profiles (MSPs), yielding 91 peaks. Subsequently, MSPs were classified according to their AMR profiles, and category labels (such as S and R) were binarized, with 0 and 1 designating MSPs as susceptible or resistant, respectively. The MSPs were randomly split into 70% training and 30% test datasets based on their binarized AMR profiles. The three distinct ML algorithms, including RF, NB, and LR were trained using a 10-fold cross-validation. Overall, classifiers identifying susceptible isolates as well as those resistant to ciprofloxacin and tetracycline exhibited overall good performance with maximum sensitivity and precision of 92.3 and 81.2%, respectively.
5.2.2. Raman spectroscopy (RS)
Raman spectroscopy is an inelastic scattering-based technology that enables real-time monitoring of the chemical composition and metabolism of a single living microbe (Rostron et al., Citation2016). Complex and high-dimensional Raman bands offer a wealth of information about distinct cell phenotypes, enabling the differentiation between diverse bacteria (Lu et al., Citation2023). Also, Raman spectroscopy is utilized to identify AMR, particularly when combined with stable isotope probing like heavy water labeling. According to theory, the label-free features of bacterial Raman spectra are effective phenotypic indicators of AMR (Lu et al., Citation2023).
Moreover, it can be easier and more efficient to identify AMR mechanisms based on the Raman spectrum when large datasets of genomes are combined with ML. Saikia et al. (Citation2022) demonstrated the ability to combine ML with Raman spectroscopy to differentiate between colistin-sensitive and resistant Escherichia coli. The AST of E. coli strains were assessed by broth microdilution method to obtain the Raman spectra. The spectra of colistin-resistant and sensitive cells were utilized as input to investigate the potential of PCA categorization. PCA was used to classify bacteria into sensitive and resistant categories based on the input data, which included control (14 spectra), 3.9 μg/mL (30 spectra), 5.0 μg/mL (28 spectra), 6.5 μg/mL (33 spectra), and 7.8 μg/mL (24 spectra). Then, it converted the variables (wavenumbers) into uncorrelated variables, also known as principal components (PCs), without changing the total variance in the spectral data. Overall, PCA of bacteria resistant to colistin indicated exclusive clusters by concentration. These findings show that it is possible to distinguish resistant cells from sensitive cells as well as resistant bacteria that are concentration-specific.
5.2.3. Fourier-transform infrared spectroscopy (FTIR)
FTIR spectroscopy is an effective technique for detecting minor molecular changes (Sharaha et al., Citation2021). This can offer precise details regarding the molecular composition of samples, especially those connected to the emergence of bacterial AMR (Suleiman et al., Citation2022). AMR in bacteria typically results from small genetic changes (mutations) in the genome (Sharaha et al., Citation2021). Therefore, the difference in infrared absorption spectra between sensitive and resistant isolates to certain antibiotics can be subtle (Suleiman et al., Citation2022). For this reason, ML is a useful technique for the classification of these databases because it can precisely differentiate even slight differences.
Recently, Sharaha et al. (Citation2021) exhibited the potential of FTIR spectroscopy combined with ML as an effective technique for a susceptibility test of Klebsiella pneumoniae. Nine distinct antibiotics were utilized to evaluate the susceptibility of 1190 isolates. They employed two classifiers, such as RF and XGBoost to distinguish between the resistant and sensitive K. pneumoniae isolates for individual-specific antibiotics. For both classifiers, they specified two hyperparameters: the maximum depth and the number of trees. In RF, the number of trees was 500–100 and the maximum depth was randomly assigned, whereas there were 900–1100 trees and a maximum depth of 5–7 in XGBoost. They trained the classifiers using both hyperparameters and sets determined by nested cross-validation, including the number of trees and maximum depth. With an accuracy rate of more than 80%, they were able to separate the isolates into sensitive or resistant to multiple antibiotics.
6. Challenges, potential strategies, and conclusions
6.1. Challenges and potential strategies
Despite promising applications of ML for the detection of AMR bacteria, several challenges should be considered in the practice application of ML. First The knowledge of ML might be limited for many researchers or engineers in the food industry. Thus, cooperation with specialists in computer science and technology, intelligent science or mathematical information is encouraged for the applications of ML. Additionally, the ML application in the food industry may face challenges associated with the need for standardized methods. The quality of training datasets cannot be guaranteed since the results from various lab groups exhibit a deviation. To ensure the consistency and comparability of data obtained from different studies, the development of standardized protocols for AMR detection is essential (Sunuwar & Azad, Citation2021). And data is rarely perfect regardless of the source, and this may introduce noise. Data cleaning and the application of normalization might be the solutions to mitigating the noise and inconsistency. This can improve the precision and efficiency of ML algorithms (Ali et al., Citation2023).
7. Conclusions
This review offers a comprehensive summary of the advancements of ML models for the surveillance of AMR in the agri-food chain. The application of ML combined with WGS and spectroscopy techniques to identify and predict AMR in foodborne pathogens is crucial to assure food safety. They are efficient in deciphering complicated data sets and analyzing enormous volumes of data, providing reliable alternatives to traditional methods. Thus, these ML applications can provide a promising strategy for tackling the AMR and, ultimately, improving global health.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Abd-Alhalem, S. M., Soliman, N. F., Eldin, S., Abd Elrahman, S. E., Ismail, N. A., El-Rabaie, E. S. M., & El-Samie, F. E. A. (2020). Bacterial classification with convolutional neural networks based on different data reduction layers. Nucleosides, Nucleotides & Nucleic Acids, 39(4), 493–16. https://doi.org/10.1080/15257770.2019.1645851
- Adnan, M. N., & Islam, M. Z. (2016). Optimizing the number of trees in a decision forest to discover a subforest with high ensemble accuracy using a genetic algorithm. Knowledge-Based Systems, 110, 86–97. https://doi.org/10.1016/j.knosys.2016.07.016
- Aires de Sousa, M. (2017). Methicillin-resistant staphylococcus aureus among animals: Current overview. Clinical Microbiology and Infection, 23(6), 373–380. https://doi.org/10.1016/j.cmi.2016.11.002
- Al-Aidaroos, K. M., Bakar, A. A., & Othman, Z. (2010, March). Naive Bayes variants in classification learning. In 2010 International Conference on Information Retrieval & Knowledge Management (CAMP) (pp. 276–281). IEEE. https://doi.org/10.1109/INFRKM.2010.5466902
- Alcock, B. P., Raphenya, A. R., Lau, T. T., Tsang, K. K., Bouchard, M., Edalatmand, A., McArthur, A. G., Nguyen, A. L. V., Cheng, A. A., Liu, S., Min, S. Y., Miroshnichenko, A., Tran, H.-K., Werfalli, R. E., Nasir, J. A., Oloni, M., Speicher, D. J., Florescu, A. … Domselaar, G. V. (2020). CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Research, 48(D1), D517–D525. https://doi.org/10.1093/nar/gkz935
- Ali, T., Ahmed, S., & Aslam, M. (2023). Artificial intelligence for antimicrobial resistance prediction: Challenges and opportunities towards practical implementation. Antibiotics, 12(3), 523. https://doi.org/10.3390/antibiotics12030523
- Anahtar, M. N., Yang, J. H., Kanjilal, S., & McAdam, A. J. (2021). Applications of machine learning to the problem of antimicrobial resistance: An emerging model for translational research. Journal of Clinical Microbiology, 59(7), 10–1128. https://doi.org/10.1128/jcm.01260-20
- Andreopoulos, W. B., Geller, A. M., Lucke, M., Balewski, J., Clum, A., Ivanova, N. N., & Levy, A. (2022). Deeplasmid: Deep learning accurately separates plasmids from bacterial chromosomes. Nucleic Acids Research, 50(3), e17. https://doi.org/10.1093/nar/gkab1115
- Anjum, M. F., Marco-Jimenez, F., Duncan, D., Marín, C., Smith, R. P., & Evans, S. J. (2019). Livestock-associated methicillin-resistant staphylococcus aureus from animals and animal products in the UK. Frontiers in Microbiology, 10, 2136. https://doi.org/10.3389/fmicb.2019.02136
- Annunziato, G. (2019). Strategies to overcome antimicrobial resistance (AMR) making use of non-essential target inhibitors: A review. International Journal of Molecular Sciences, 20(23), 5844. https://doi.org/10.3390/ijms20235844
- Arredondo-Alonso, S., Rogers, M. R. C., Braat, J. C., Verschuuren, T. D., Top, J., Corander, J., Willems, R. J. L., & Schürch, A. C. (2018). Mlplasmids: A user-friendly tool to predict plasmid-and chromosome-derived sequences for single species. Microbial Genomics, 4(11). https://doi.org/10.1099/mgen.0.000224
- Asselman, A., Khaldi, M., & Aammou, S. (2023). Enhancing the prediction of student performance based on the machine learning XGBoost algorithm. Interactive Learning Environments, 31(6), 3360–3379. https://doi.org/10.1080/10494820.2021.1928235
- Awad, M., Khanna, R., Awad, M., & Khanna, R. (2015). Support vector machines for classification. Efficient Learning Machines: Theories, Concepts, and Applications for Engineers and System Designers, 39–66. https://doi.org/10.1007/978-1-4302-5990-9_3
- Azmi, S. S., & Baliga, S. (2020). An Overview of Boosting Decision Tree Algorithms utilizing AdaBoost and XGBoost Boosting strategies. International Research Journal of Engineering & Technology, 7(5). 6867–6870
- Baker, M., Zhang, X., Maciel-Guerra, A., Dong, Y., Wang, W., Hu, Y., Renney, D., Hu, Y., Liu, L., Li, H., Tong, Z., Zhang, M., Geng, Y., Zhao, L., Hao, Z., Senin, N., Chen, J., Peng, Z., Li, F., & Dottorini, T. (2023). Machine learning and metagenomics reveal shared antimicrobial resistance profiles across multiple chicken farms and abattoirs in China. Nature Food, 4(8), 707–720. https://doi.org/10.1038/s43016-023-00814-w
- Bandyopadhyay, S., & Samanta, I. (2020). Antimicrobial resistance in agri-food chain and companion animals as a re-emerging menace in post-COVID epoch: Low-and middle-income countries perspective and mitigation strategies. Frontiers in Veterinary Science, 7, 620. https://doi.org/10.3389/fvets.2020.00620
- Beceiro, A., Tomás, M., & Bou, G. (2013). Antimicrobial resistance and virulence: A successful or deleterious association in the bacterial world? Clinical Microbiology Reviews, 26(2), 185–230. https://doi.org/10.1128/cmr.00059-12
- Bintsis, T. (2017). Foodborne pathogens. AIMS Microbiology, 3(3), 529. https://doi.org/10.3934/microbiol.2017.3.529
- Blancquaert, D., Storozhenko, S., Loizeau, K., De Steur, H., De Brouwer, V., Viaene, J., Ravanel, S., Rébeillé, F., Lambert, W., & Van Der Straeten, D. (2010). Folates and folic acid: From fundamental research toward sustainable health. Critical Reviews in Plant Science, 29(1), 14–35. https://doi.org/10.1080/07352680903436283
- Bonomo, R. A. (2017). β-lactamases: A focus on current challenges. Cold Spring Harbor Perspectives in Medicine, 7(1), a025239. https://doi.org/10.1101/cshperspect.a025239
- Bonvegna, M., Tomassone, L., Christensen, H., & Olsen, J. E. (2022). Whole genome sequencing (WGS) analysis of virulence and AMR genes in extended-spectrum β-lactamase (ESBL)-producing Escherichia coli from animal and Environmental Samples in four Italian swine farms. Antibiotics, 11(12), 1774. https://doi.org/10.3390/antibiotics11121774
- Boolchandani, M., D’Souza, A. W., & Dantas, G. (2019). Sequencing-based methods and resources to study antimicrobial resistance. Nature Reviews Genetics, 20(6), 356–370. https://doi.org/10.1038/s41576-019-0108-4
- Breijyeh, Z., Jubeh, B., & Karaman, R. (2020). Resistance of gram-negative bacteria to current antibacterial agents and approaches to resolve it. Molecules, 25(6), 1340. https://doi.org/10.3390/molecules25061340
- Breiman, L. (2001). Random forests. Machine Learning, 45(1), 5–32. https://doi.org/10.1023/A:1010933404324
- Bush, K. (2010). Alarming β-lactamase-mediated resistance in multidrug-resistant Enterobacteriaceae. Current Opinion in Microbiology, 13(5), 558–564. https://doi.org/10.1016/j.mib.2010.09.006
- Chauhan, V. K., Dahiya, K., & Sharma, A. (2019). Problem formulations and solvers in linear SVM: A review. Artificial Intelligence Review, 52(2), 803–855. https://doi.org/10.1007/s10462-018-9614-6
- Chen, Q., Lan, C., Zhao, L., Wang, J., Chen, B., & Chen, Y. P. P. (2017). Recent advances in sequence assembly: Principles and applications. Briefings in Functional Genomics, 16(6), 361–378. https://doi.org/10.1093/bfgp/elx006
- Chen, C., & Wu, F. (2021). Livestock-associated methicillin-resistant Staphylococcus aureus (LA-MRSA) colonisation and infection among livestock workers and veterinarians: A systematic review and meta-analysis. Occupational and Environmental Medicine, 78(7), 530–540. https://doi.org/10.1136/oemed-2020-106418
- Chin, F. Y., Leung, H. C., & Yiu, S. M. (2014). Sequence assembly using next generation sequencing data—challenges and solutions. Science China Life Sciences, 57(11), 1140–1148. https://doi.org/10.1007/s11427-014-4752-9
- Chuang, Y. Y., & Huang, Y. C. (2015). Livestock-associated meticillin-resistant staphylococcus aureus in Asia: An emerging issue? International Journal of Antimicrobial Agents, 45(4), 334–340. https://doi.org/10.1016/j.ijantimicag.2014.12.007
- Crespo-Piazuelo, D., & Lawlor, P. G. (2021). Livestock-associated methicillin-resistant staphylococcus aureus (LA-MRSA) prevalence in humans in close contact with animals and measures to reduce on-farm colonisation. Irish Veterinary Journal, 74(1), 1–12. https://doi.org/10.1186/s13620-021-00200-7
- Davis, J. J., Wattam, A. R., Aziz, R. K., Brettin, T., Butler, R., Butler, R. M., Stevens, R., Conrad, N., Dickerman, A., Dietrich, E. M., Gabbard, J. L., Gerdes, S., Guard, A., Kenyon, R. W., Machi, D., Mao, C., Murphy-Olson, D., Nguyen, M. … Yoo, H. (2020). The PATRIC bioinformatics resource center: Expanding data and analysis capabilities. Nucleic Acids Research, 48(D1), D606–D612. https://doi.org/10.1093/nar/gkz943
- De Boer, E., Zwartkruis-Nahuis, J. T. M., Wit, B., Huijsdens, X. W., De Neeling, A. J., Bosch, T., Van Oosterom, R. A. A., Vila, A., & Heuvelink, A. E. (2009). Prevalence of methicillin-resistant Staphylococcus aureus in meat. International Journal of Food Microbiology, 134(1–2), 52–56. https://doi.org/10.1016/j.ijfoodmicro.2008.12.007
- Delcour, A. H. (2009). Outer membrane permeability and antibiotic resistance. Biochimica Et Biophysica Acta (BBA)-Proteins and Proteomics, 1794(5), 808–816. https://doi.org/10.1016/j.bbapap.2008.11.005
- Dhanda, G., Acharya, Y., & Haldar, J. (2023). Antibiotic adjuvants: A versatile approach to combat antibiotic resistance. American Chemical Society Omega, 8(12), 10757–10783. https://doi.org/10.1021/acsomega.3c00312
- Dieckmann, R., Hammerl, J. A., Hahmann, H., Wicke, A., Kleta, S., Dabrowski, P. W., Nitsche, A., Stämmler, M., Al Dahouk, S., & Lasch, P. (2016). Rapid characterisation of Klebsiella oxytoca isolates from contaminated liquid hand soap using mass spectrometry, FTIR and Raman spectroscopy. Faraday Discussions, 187, 353–375. https://doi.org/10.1039/C5FD00165J
- Doyle, M. E. (2015). Multidrug-resistant pathogens in the food supply. Foodborne Pathogens and Disease, 12(4), 261–279. https://doi.org/10.1089/fpd.2014.1865
- Du, T. Y. (2019). Dimensionality reduction techniques for visualizing morphometric data: Comparing principal component analysis to nonlinear methods. Evolutionary Biology, 46(1), 106–121. https://doi.org/10.1007/s11692-018-9464-9
- Du, D., Wang-Kan, X., Neuberger, A., Van Veen, H. W., Pos, K. M., Piddock, L. J., & Luisi, B. F. (2018). Multidrug efflux pumps: Structure, function and regulation. Nature Reviews Microbiology, 16(9), 523–539. https://doi.org/10.1038/s41579-018-0048-6
- Economou, V., & Gousia, P. (2015). Agriculture and food animals as a source of antimicrobial-resistant bacteria. Infection and Drug Resistance, 49–61. https://doi.org/10.2147/IDR.S55778
- EFSA & ECDC. (2022). The European Union summary report on antimicrobial resistance in zoonotic and indicator bacteria from humans, animals and food in 2019-2020. European Food Safety Authority Journal, 20(3), e07209. https://doi.org/10.2903/j.efsa.2022.7209
- Ehuwa, O., Jaiswal, A. K., & Jaiswal, S. (2021). Salmonella, food safety and food handling practices. Foods, 10(5), 907. https://doi.org/10.3390/foods10050907
- El Bouchefry, K., & de Souza, R. S. (2020). Learning in big data: Introduction to machine learning. Knowledge Discovery in Big Data from Astronomy and Earth Observation, 225–249. https://doi.org/10.1016/B978-0-12-819154-5.00023-0
- Esener, N., Maciel-Guerra, A., Giebel, K., Lea, D., Green, M. J., Bradley, A. J., Dottorini, T., & Holmes, M. (2021). Mass spectrometry and machine learning for the accurate diagnosis of benzylpenicillin and multidrug resistance of staphylococcus aureus in bovine mastitis. PLoS Computational Biology, 17(6), e1009108. https://doi.org/10.1371/journal.pcbi.1009108
- Espinoza, N., Rojas, J., Pollett, S., Meza, R., Patino, L., Leiva, M., Camiña, M., Bernal, M., Reynolds, N. D., Maves, R., Tilley, D. H., Kasper, M., & Simons, M. P. (2020). Validation of the T86I mutation in the gyrA gene as a highly reliable real time PCR target to detect Fluoroquinolone-resistant Campylobacter jejuni. BMC Infectious Diseases, 20(1), 1–7. https://doi.org/10.1186/s12879-020-05202-4
- Feldgarden, M., Brover, V., Haft, D. H., Prasad, A. B., Slotta, D. J., Tolstoy, I., Tyson, G. H., Zhao, S., Hsu, C.-H., McDermott, P. F., Tadesse, D. A., Morales, C., Simmons, M., Tillman, G., Wasilenko, J., Folster, J. P., & Klimke, W. (2019). Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrobial Agents and Chemotherapy, 63(11), 10–1128. https://doi.org/10.1128/aac.00483-19
- Feucherolles, M., Nennig, M., Becker, S. L., Martiny, D., Losch, S., Penny, C., Cauchie, H.-M., & Ragimbeau, C. (2022). Combination of MALDI-TOF mass spectrometry and machine learning for rapid antimicrobial resistance screening: The case of Campylobacter spp. Frontiers in Microbiology, 12, 804484. https://doi.org/10.3389/fmicb.2021.804484
- Florensa, A. F., Kaas, R. S., Clausen, P. T. L. C., Aytan-Aktug, D., & Aarestrup, F. M. (2022). ResFinder–an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microbial Genomics, 8(1). https://doi.org/10.1099/mgen.0.000748
- Frasao, B. D. S., Medeiros, V., Barbosa, A. V., De Aguiar, W. S., Dos Santos, F. F., Abreu, D. L. D. C., De Aquino, M. H. C., & de Aquino, M. H. C. (2015). Detection of fluoroquinolone resistance by mutation in gyrA gene of Campylobacter spp. isolates from broiler and laying (Gallus gallus domesticus) hens, from Rio de Janeiro State, Brazil. Ciencia Rural, 45(11), 2013–2018. https://doi.org/10.1590/0103-8478cr20141712
- Freeland, G., Hettiarachchy, N., Atungulu, G. G., Apple, J., & Mukherjee, S. (2023). Strategies to combat antimicrobial resistance from farm to table. Food Reviews International, 39(1), 27–40. https://doi.org/10.1080/87559129.2021.1893744
- Gan, T., Shu, G., Fu, H., Yan, Q., Zhang, W., Tang, H., Yin, L., Zhao, L., & Lin, J. (2021). Antimicrobial resistance and genotyping of Staphylococcus aureus obtained from food animals in Sichuan Province, China. BMC Veterinary Research, 17(1), 177. https://doi.org/10.1186/s12917-021-02884-z
- Giedraitienė, A., Vitkauskienė, A., Naginienė, R., & Pavilonis, A. (2011). Antibiotic resistance mechanisms of clinically important bacteria. Medicina, 47(3), 19. https://doi.org/10.3390/medicina47030019
- Golding, G. R., Bryden, L., Levett, P. N., McDonald, R. R., Wong, A., Wylie, J., Graham, M. R., Tyler, S., Van Domselaar, G., Simor, A. E., Gravel, D., & Mulvey, M. R. (2010). Livestock-associated methicillin-resistant Staphylococcus aureus sequence type 398 in humans, Canada. Emerging Infectious Diseases, 16(4), 587. https://doi.org/10.3201/eid1604.091435
- Grudlewska-Buda, K., Bauza-Kaszewska, J., Wiktorczyk-Kapischke, N., Budzyńska, A., Gospodarek-Komkowska, E., & Skowron, K. (2023). Antibiotic resistance in selected emerging bacterial foodborne pathogens-an issue of concern? Antibiotics, 12(5), 880. https://doi.org/10.3390/antibiotics12050880
- Gupta, S. K., Padmanabhan, B. R., Diene, S. M., Lopez-Rojas, R., Kempf, M., Landraud, L., & Rolain, J. M. (2014). ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrobial Agents and Chemotherapy, 58(1), 212–220. https://doi.org/10.1128/AAC.01310-13
- Gurevich, A., Saveliev, V., Vyahhi, N., & Tesler, G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics, 29(8), 1072–1075. https://doi.org/10.1093/bioinformatics/btt086
- Haag, A. F., Fitzgerald, J. R., Penadés, J. R., Fischetti, V. A., Novick, R. P., Ferretti, J. J., Portnoy, D. A., Braunstein, M., & Rood, J. I. (2019). Staphylococcus aureus in Animals. Microbiology Spectrum, 7(3), 10–1128. https://doi.org/10.1128/microbiolspec.gpp3-0060-2019
- Hatje, K., & Kollmar, M. (2012). A phylogenetic analysis of the brassicales clade based on an alignment-free sequence comparison method. Frontiers in Plant Science, 3, 192. https://doi.org/10.3389/fpls.2012.00192
- Hosain, M. Z., Kabir, S. L., & Kamal, M. M. (2021). Antimicrobial uses for livestock production in developing countries. Veterinary World, 14(1), 210. https://doi.org/10.14202/vetworld.2021.210-221
- Hou, T. Y., Chiang-Ni, C., & Teng, S. H. (2019). Current status of MALDI-TOF mass spectrometry in clinical microbiology. Journal of Food and Drug Analysis, 27(2), 404–414. https://doi.org/10.1016/j.jfda.2019.01.001
- Hudson, J. A., Frewer, L. J., Jones, G., Brereton, P. A., Whittingham, M. J., & Stewart, G. (2017). The agri-food chain and antimicrobial resistance: A review. Trends in Food Science & Technology, 69, 131–147. https://doi.org/10.1016/j.tifs.2017.09.007
- Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., McArthur, A. G., Dave, B. M., Pereira, S., Sharma, A. N., Doshi, S., Courtot, M., Lo, R., Williams, L. E., Frye, J. G., Elsayegh, T., Sardar, D., Westman, E. L., & Wright, G. D. (2017). CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Research, 45(D1), D566–573. https://doi.org/10.1093/nar/gkw1004
- Kapoor, G., Saigal, S., & Elongavan, A. (2017). Action and resistance mechanisms of antibiotics: A guide for clinicians. Journal of Anaesthesiology, Clinical Pharmacology, 33(3), 300. https://doi.org/10.4103/joacp.JOACP_349_15
- Khademi, F., Sahebkar, A., & Chaves Lopez, C. (2020). Prevalence of fluoroquinolone-resistant Campylobacter species in Iran: A systematic review and meta-analysis. International Journal of Microbiology, 2020, 1–14. https://doi.org/10.1155/2020/8868197
- Khanna, T., Friendship, R., Dewey, C., & Weese, J. S. (2008). Methicillin resistant staphylococcus aureus colonization in pigs and pig farmers. Veterinary Microbiology, 128(3–4), 298–303. https://doi.org/10.1016/j.vetmic.2007.10.006
- Khezri, A., Avershina, E., & Ahmad, R. (2021). Hybrid assembly provides improved resolution of plasmids, antimicrobial resistance genes, and virulence factors in Escherichia coli and Klebsiella pneumoniae clinical isolates. Microorganisms [Internet], 9(12), 2560. https://doi.org/10.3390/microorganisms9122560
- Koch, B. J., Hungate, B. A., & Price, L. B. (2017). Food‐animal production and the spread of antibiotic resistance: The role of ecology. Frontiers in Ecology and the Environment, 15(6), 309–318. https://doi.org/10.1002/fee.1505
- Kourou, K., Exarchos, T. P., Exarchos, K. P., Karamouzis, M. V., & Fotiadis, D. I. (2015). Machine learning applications in cancer prognosis and prediction. Computational and Structural Biotechnology Journal, 13, 8–17. https://doi.org/10.1016/j.csbj.2014.11.005
- Larsen, J., Raisen, C. L., Ba, X., Sadgrove, N. J., Padilla-González, G. F., Simmonds, M. S., Larsen, A. R., Kerschner, H., Apfalter, P., Hartl, R., Deplano, A., Vandendriessche, S., Černá Bolfíková, B., Hulva, P., Arendrup, M. C., Hare, R. K., Barnadas, C., Stegger, M. … Harrison, E. M. (2022). Emergence of methicillin resistance predates the clinical use of antibiotics. Nature, 602(7895), 135–141. https://doi.org/10.1038/s41586-021-04265-w
- Liu, Z., Deng, D., Lu, H., Sun, J., Lv, L., Li, S., Peng, G., Ma, X., Li, J., Li, Z., Rong, T., & Wang, G. (2020). Evaluation of machine learning models for predicting antimicrobial resistance of actinobacillus pleuropneumoniae from whole genome sequences. Frontiers in Microbiology, 11, 48. https://doi.org/10.3389/fmicb.2020.00048
- Livermore, D. M. (2000). Antibiotic resistance in staphylococci. International Journal of Antimicrobial Agents, 16, 3–10. https://doi.org/10.1016/S0924-8579(00)00299-5
- Lu, W., Li, H., Qiu, H., Wang, L., Feng, J., & Fu, Y. V. (2023). Identification of pathogens and detection of antibiotic susceptibility at single-cell resolution by Raman spectroscopy combined with machine learning. Frontiers in Microbiology, 13, 1076965. https://doi.org/10.3389/fmicb.2022.1076965
- Maguire, F., Rehman, M. A., Carrillo, C., Diarra, M. S., Beiko, R. G., & Gilbert, J. A. (2019). Identification of primary antimicrobial resistance drivers in agricultural nontyphoidal Salmonella enterica serovars by using machine learning. mSystems [Internet], 4(4), 10–1128. https://doi.org/10.1128/msystems.00211-19
- Mann, A., Nehra, K., Rana, J. S., & Dahiya, T. (2021). Antibiotic resistance in agriculture: Perspectives on upcoming strategies to overcome upsurge in resistance. Current Research in Microbial Sciences, 2, 100030. https://doi.org/10.1016/j.crmicr.2021.100030
- McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., Wright, G. D., Canova, M. J., De Pascale, G., Ejim, L., Kalan, L., King, A. M., Koteva, K., Morar, M., Mulvey, M. R., O’Brien, J. S., Pawlowski, A. C., Piddock, L. J. V. … Yu, T. (2013). The comprehensive antibiotic resistance database. Antimicrobial Agents and Chemotherapy, 57(7), 3348–3357. https://doi.org/10.1128/aac.00419-13
- McEwen, S. A., & Fedorka-Cray, P. J. (2002). Antimicrobial use and resistance in animals. Clinical Infectious Diseases, 34(Supplement_3), S93–S106. https://doi.org/10.1086/340246
- Medalla, F., Gu, W., Friedman, C. R., Judd, M., Folster, J., Griffin, P. M., & Hoekstra, R. M. (2021). Increased incidence of antimicrobial-resistant nontyphoidal Salmonella infections, United States, 2004-2016. Emerging Infectious Diseases, 27(6), 1662. https://doi.org/10.3201/eid2706.204486
- Mulchandani, R., Wang, Y., Gilbert, M., Van Boeckel, T. P., & Odetokun, I. A. (2023). Global trends in antimicrobial use in food-producing animals: 2020 to 2030. PLoS Global Public Health, 3(2), e0001305. https://doi.org/10.1371/journal.pgph.0001305
- Munita, J. M., & Arias, C. A. (2016). Mechanisms of antibiotic resistance. Virulence Mechanisms of Bacterial Pathogens, 481–511. https://doi.org/10.1128/microbiolspec.vmbf-0016-2015
- Nelson, J. M., Chiller, T. M., Powers, J. H., & Angulo, F. J. (2007). Fluoroquinolone-resistant Campylobacter species and the withdrawal of fluoroquinolones from use in poultry: A public health success story. Clinical Infectious Diseases, 44(7), 977–980. https://doi.org/10.1086/512369
- Nguyen, M., Brettin, T., Long, S. W., Musser, J. M., Olsen, R. J., Olson, R., Shukla, M., Stevens, R. L., Xia, F., Yoo, H., & Davis, J. J. (2018). Developing an in silico minimum inhibitory concentration panel test for Klebsiella pneumoniae. Scientific Reports, 8(1), 421. https://doi.org/10.1038/s41598-017-18972-w
- Nishino, K., Yamasaki, S., Nakashima, R., Zwama, M., & Hayashi-Nishino, M. (2021). Function and inhibitory mechanisms of multidrug efflux pumps. Frontiers in Microbiology, 12, 737288. https://doi.org/10.3389/fmicb.2021.737288
- Olson, R. D., Assaf, R., Brettin, T., Conrad, N., Cucinell, C., Davis, J. J., Dempsey, D. M., Dickerman, A., Dietrich, E. M., Kenyon, R. W., Kuscuoglu, M., Lefkowitz, E. J., Lu, J., Machi, D., Macken, C., Mao, C., Niewiadomska, A., Nguyen, M., Olsen, G. J., … Stevens, R. L. (2023). Introducing the bacterial and viral bioinformatics resource center (BV-BRC): A resource combining PATRIC, IRD and ViPR. Nucleic Acids Research, 51(D1), D678–D689. https://doi.org/10.1093/nar/gkac1003
- Papp, M., & Solymosi, N. (2022). Review and comparison of antimicrobial resistance gene databases. Antibiotics, 11(3), 339. https://doi.org/10.3390/antibiotics11030339
- Peng, Z., Maciel-Guerra, A., Baker, M., Zhang, X., Hu, Y., Wang, W., Rong, J., Zhang, J., Xue, N., Barrow, P., Renney, D., Stekel, D., Williams, P., Liu, L., Chen, J., Li, F., & Dottorini, T. (2022). Whole-genome sequencing and gene sharing network analysis powered by machine learning identifies antibiotic resistance sharing between animals, humans and environment in livestock farming. PloS Computational Biology, 18(3), e1010018. https://doi.org/10.1371/journal.pcbi.1010018
- Ramtahal, M. A., Amoako, D. G., Akebe, A. L., Somboro, A. M., Bester, L. A., & Essack, S. Y. (2022). A public health insight into Salmonella in poultry in Africa: A review of the past Decade: 2010-2020. Microbial Drug Resist, 28(6), 710–733. https://doi.org/10.1089/mdr.2021.0384
- Ren, Y., Chakraborty, T., Doijad, S., Falgenhauer, L., Falgenhauer, J., Goesmann, A., Hauschild, A.-C., Schwengers, O., & Heider, D. (2022). Prediction of antimicrobial resistance based on whole-genome sequencing and machine learning. Bioinformatics, 38(2), 325–334. https://doi.org/10.1093/bioinformatics/btab681
- Ren, J., Lee, S. D., Chen, X., Kao, B., Cheng, R., & Cheung, D. (2009, December). Naive Bayes classification of uncertain data. In 2009 Ninth IEEE International Conference on Data Mining (pp. 944–949). IEEE. https://doi.org/10.1109/ICDM.2009.90
- Reygaert, W. C. (2018). An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiology, 4(3), 482. https://doi.org/10.3934/microbiol.2018.3.482
- Rodrigues, G. L., Panzenhagen, P., Ferrari, R. R., Dos Santos, A., Paschoalin, V. M. F., & Conte-Junior, C. A. (2020). Frequency of antimicrobial resistance genes in Salmonella from Brazil by in silico whole-genome sequencing analysis: An overview of the last four decades. Frontiers in Microbiology, 11, 1864. https://doi.org/10.3389/fmicb.2020.01864
- Rokney, A., Valinsky, L., Vranckx, K., Feldman, N., Agmon, V., Moran-Gilad, J., & Weinberger, M. (2020). WGS-based prediction and analysis of antimicrobial resistance in campylobacter jejuni isolates from Israel. Frontiers in Cellular and Infection Microbiology, 10, 365. https://doi.org/10.3389/fcimb.2020.00365
- Rostron, P., Gaber, S., & Gaber, D. (2016). Raman spectroscopy, review. Laser, 6(1), 50–64.
- Saikia, D., Jadhav, P., Hole, A. R., Krishna, C. M., & Singh, S. P. (2022). Unraveling the secrets of colistin resistance with label-free raman spectroscopy. Biosensors, 12(9), 749. https://doi.org/10.3390/bios12090749
- Salam, M. A., Al-Amin, M. Y., Pawar, J. S., Akhter, N., & Lucy, I. B. (2023). Conventional methods and future trends in antimicrobial susceptibility testing. Saudi Journal of Biological Sciences, 30(3), 103582. https://doi.org/10.1016/j.sjbs.2023.103582
- Samtiya, M., Matthews, K. R., Dhewa, T., & Puniya, A. K. (2022). Antimicrobial resistance in the food chain: Trends, mechanisms, pathways, and possible regulation strategies. Foods, 11(19), 2966. https://doi.org/10.3390/foods11192966
- Sarker, I. H. (2021). Machine learning: Algorithms, real-world applications and research directions. SN Computer Science, 2(3), 160. https://doi.org/10.1007/s42979-021-00592-x
- Schmitt, J., Bönig, J., Borggräfe, T., Beitinger, G., & Deuse, J. (2020). Predictive model-based quality inspection using machine learning and edge cloud computing. Advanced Engineering Informatics, 45, 101101. https://doi.org/10.1016/j.aei.2020.101101
- Sharaha, U., Suleiman, M., Abu-Aqil, G., Riesenberg, K., Lapidot, I., Salman, A., & Huleihel, M. (2021). Determination of Klebsiella pneumoniae susceptibility to antibiotics using infrared microscopy. Analytical Chemistry, 93(40), 13426–13433. https://doi.org/10.1021/acs.analchem.1c00734
- Sinaga, K. P., & Yang, M. S. (2020). Unsupervised K-means clustering algorithm. IEEE Access, 8, 80716–80727. https://doi.org/10.1109/ACCESS.2020.2988796
- Smith, T. C., Male, M. J., Harper, A. L., Kroeger, J. S., Tinkler, G. P., Moritz, E. D., Capuano, A. W., Herwaldt, L. A., & Diekema, D. J. (2009). Methicillin-resistant Staphylococcus aureus (MRSA) strain ST398 is present in midwestern U.S. Swine and swine workers. Public Library of Science One, 4(1), e4258. https://doi.org/10.1371/journal.pone.0004258
- Song, Y. Y., & Ying, L. U. (2015). Decision tree methods: Applications for classification and prediction. Shanghai Archives of Psychiatry, 27(2), 130. https://doi.org/10.11919/j.issn.1002-0829.215044
- Spänig, S., & Heider, D. (2019). Encodings and models for antimicrobial peptide classification for multi-resistant pathogens. BioData Mining, 12(1), 1–29. https://doi.org/10.1186/s13040-019-0196-x
- Stein, L. (2001). Genome annotation: From sequence to biology. Nature Reviews Genetics, 2(7), 493–503. https://doi.org/10.1038/35080529
- Stratev, D., & Odeyemi, O. A. (2016). Antimicrobial resistance of Aeromonas hydrophila isolated from different food sources: A mini-review. Journal of Infection and Public Health, 9(5), 535–544. https://doi.org/10.1016/j.jiph.2015.10.006
- Suleiman, M., Abu-Aqil, G., Sharaha, U., Riesenberg, K., Lapidot, I., Salman, A., & Huleihel, M. (2022). Infra-red spectroscopy combined with machine learning algorithms enables early determination of Pseudomonas aeruginosa’s susceptibility to antibiotics. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 274, 121080. https://doi.org/10.1016/j.saa.2022.121080
- Sun, S., Selmer, M., Andersson, D. I., & Cloeckaert, A. (2014). Resistance to β-lactam antibiotics conferred by point mutations in penicillin-binding proteins PBP3, PBP4 and PBP6 in Salmonella enterica. Public Library of Science ONE, 9(5), e97202. https://doi.org/10.1371/journal.pone.0097202
- Sunuwar, J., & Azad, R. K. (2021). A machine learning framework to predict antibiotic resistance traits and yet unknown genes underlying resistance to specific antibiotics in bacterial strains. Briefings in Bioinformatics, 22(6), bbab179. https://doi.org/10.1093/bib/bbab179
- Tang, X., Shang, J., Ji, Y., & Sun, Y. (2023). Plasme: A tool to identify PLASMid contigs from short-read assemblies using transformer. Nucleic Acids Research, 51(15), e83–e83. https://doi.org/10.1093/nar/gkad578
- Turner, N. A., Sharma-Kuinkel, B. K., Maskarinec, S. A., Eichenberger, E. M., Shah, P. P., Holland, T. L., & Fowler Jr, V. G. (2019). Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nature Reviews Microbiology, 17(4), 203–218. https://doi.org/10.1038/s41579-018-0147-4
- Uddin, T. M., Chakraborty, A. J., Khusro, A., Zidan, B. R. M., Mitra, S., Emran, T. B., Dhama, K., Ripon, M. K. H., Gajdács, M., Sahibzada, M. U. K., Hossain, M. J., & Koirala, N. (2021). Antibiotic resistance in microbes: History, mechanisms, therapeutic strategies and future prospects. Journal of Infection and Public Health, 14(12), 1750–1766. https://doi.org/10.1016/j.jiph.2021.10.020
- Uelze, L., Grützke, J., Borowiak, M., Hammerl, J. A., Juraschek, K., Deneke, C., Tausch, S. H., & Malorny, B. (2020). Typing methods based on whole genome sequencing data. One Health Outlook, 2(1), 1–19. https://doi.org/10.1186/s42522-020-0010-1
- UN. (2020). Antimicrobial resistance: a global threat. UNEP - UN Environment Programme. http://www.unep.org/explore-topics/chemicals-waste/what-we-do/emerging-issues/antimicrobial-resistance-global-threat
- Urban-Chmiel, R., Marek, A., Stępień-Pyśniak, D., Wieczorek, K., Dec, M., Nowaczek, A., & Osek, J. (2022). Antibiotic resistance in bacteria-A review. Antibiotics, 11(8), 1079. https://doi.org/10.3390/antibiotics11081079
- Van Boeckel, T. P., Brower, C., Gilbert, M., Grenfell, B. T., Levin, S. A., Robinson, T. P., Teillant, A., & Laxminarayan, R. (2015). Global trends in antimicrobial use in food animals. Proceedings of the National Academy of Sciences, 112(18), 5649–5654. https://doi.org/10.1073/pnas.1503141112
- Van Boeckel, T. P., Glennon, E. E., Chen, D., Gilbert, M., Robinson, T. P., Grenfell, B. T., Levin, S. A., Bonhoeffer, S., & Laxminarayan, R. (2017). Reducing antimicrobial use in food animals. Science, 357(6358), 1350–1352. https://doi.org/10.1126/science.aao1495
- Veltcheva, D., Colles, F. M., Varga, M., Maiden, M. C., & Bonsall, M. B. (2022). Emerging patterns of fluoroquinolone resistance in Campylobacter jejuni in the UK [1998–2018]. Microbial Genomics, 8(9). https://doi.org/10.1099/mgen.0.000875
- Vinayamohan, P. G., Pellissery, A. J., & Venkitanarayanan, K. (2022). Role of horizontal gene transfer in the dissemination of antimicrobial resistance in food animal production. Current Opinion in Food Science, 47, 100882. https://doi.org/10.1016/j.cofs.2022.100882
- Wang, W., Baker, M., Hu, Y., Xu, J., Yang, D., Maciel-Guerra, A., Xue, N., Li, H., Yan, S., Li, M., Bai, Y., Dong, Y., Peng, Z., Ma, J., Li, F., & Dottorini, T. (2021). Whole-genome sequencing and machine learning analysis of Staphylococcus aureus from multiple heterogeneous sources in China reveals common genetic traits of antimicrobial resistance. mSystems [Internet], 6(3), e01185–20. https://doi.org/10.1128/msystems.01185-20
- Wang, X. L., Li, L., Li, S. M., Huang, J. Y., Fan, Y. P., Yao, Z. J., Ye, X. H., & Chen, S. D. (2017). Phenotypic and molecular characteristics of Staphylococcus aureus and methicillin-resistant Staphylococcus aureus in slaughterhouse pig-related workers and control workers in Guangdong Province, China. Epidemiology & Infection, 145(9), 1843–1851. https://doi.org/10.1017/S0950268817000085
- WHO. (2017a). Stop using antibiotics in healthy animals to preserve their effectiveness. https://www.who.int/news/item/07-11-2017-stop-using-antibiotics-in-healthy-animals-to-prevent-the-spread-of-antibiotic-resistance
- WHO. (2017b). WHO publishes list of bacteria for which new antibiotics are urgently needed. https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed
- WHO. (2019a). New report calls for urgent action to avert antimicrobial resistance crisis. https://www.who.int/news/item/29-04-2019-new-report-calls-for-urgent-action-to-avert-antimicrobial-resistance-crisis
- WHO. (2019b). UN interagency coordination group on antimicrobial resistance presents its report to the UN SG. https://www.who.int/news/item/28-04-2019-un-interagency-coordination-group-on-antimicrobial-resistance-presents-its-report-to-the-un-sg
- Wick, R. R., Judd, L. M., Gorrie, C. L., & Holt, K. E. (2017). Completing bacterial genome assemblies with multiplex MinION sequencing. Microbial Genomics, 3(10). https://doi.org/10.1099/mgen.0.000132
- Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E., & Phillippy, A. M. (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Computational Biology, 13(6), e1005595. https://doi.org/10.1371/journal.pcbi.1005595
- Wieczorek, K., & Osek, J. (2013). Antimicrobial resistance mechanisms among Campylobacter. BioMed Research International, 2013, 1–12. https://doi.org/10.1155/2013/340605
- Xie, R., Li, J., Wang, J., Dai, W., Leier, A., Marquez-Lago, T. T., Akutsu, T., Lithgow, T., Song, J., & Zhang, Y. (2021). DeepVF: A deep learning-based hybrid framework for identifying virulence factors using the stacking strategy. Briefings in Bioinformatics, 22(3), bbaa125. https://doi.org/10.1093/bib/bbaa125
- Xiong, Z., Cui, Y., Liu, Z., Zhao, Y., Hu, M., & Hu, J. (2020). Evaluating explorative prediction power of machine learning algorithms for materials discovery using k-fold forward cross-validation. Computational Materials Science, 171, 109203. https://doi.org/10.1016/j.commatsci.2019.109203
- Yadav, S., & Kapley, A. (2021). Antibiotic resistance: Global health crisis and metagenomics. Biotechnology Reports, 29, e00604. https://doi.org/10.1016/j.btre.2021.e00604
- Yadav, S., & Shukla, S. (2016, February). Analysis of k-fold cross-validation over hold-out validation on colossal datasets for quality classification. In 2016 IEEE 6th International Conference on Advanced Computing (IACC). IEEE. https://doi.org/10.1109/IACC.2016.25
- Yang, A., Zhang, W., Wang, J., Yang, K., Han, Y., & Zhang, L. (2020). Review on the application of machine learning algorithms in the sequence data mining of DNA. Frontiers in Bioengineering and Biotechnology, 8, 1032. https://doi.org/10.3389/fbioe.2020.01032
- Yasir, M., Karim, A. M., Malik, S. K., Bajaffer, A. A., & Azhar, E. I. (2022). Application of decision-tree-based machine learning algorithms for prediction of antimicrobial resistance. Antibiotics, 11(11), 1593. https://doi.org/10.3390/antibiotics11111593
- Yuan, J., Tang, F., Qi, Z., & Zhao, H. (2023). Prediction and determination of mildew grade in grain storage based on FOA-SVM algorithm. Food Quality and Safety, 7, 7. https://doi.org/10.1093/fqsafe/fyac071
- Zhang, Z., & Castelló, A. (2017). Principal components analysis in clinical studies. Annals of Translational Medicine, 5(17), 351–351. https://doi.org/10.21037/atm.2017.07.12
- Zhang, Q., Shen, Z., & Huang, D. S. (2019). Modeling in-vivo protein-DNA binding by combining multiple-instance learning with a hybrid deep neural network. Scientific Reports, 9(1), 8484. https://doi.org/10.1038/s41598-019-44966-x
- Zhu, Q., Gao, S., Xiao, B., He, Z., Hu, S., & Yin, Y. (2023). Plasmer: An accurate and sensitive bacterial plasmid prediction tool based on machine learning of shared k-mers and genomic features. Microbiology Spectrum, 11(3), e04645–22. https://doi.org/10.1128/spectrum.04645-22