
ABSTRACT
Human respiratory syncytial virus (HRSV) accounts for the majority of lower respiratory tract infections during infancy and childhood and is associated with significant morbidity and mortality. HRSV provokes a proliferation arrest and characteristic syncytia in cellular systems such as immortalized epithelial cells. We show here that HRSV induces the expression of DNA damage markers and proliferation arrest such as P-TP53, P-ATM, CDKN1A and γH2AFX in cultured cells secondary to the production of mitochondrial reactive oxygen species (ROS). The DNA damage foci contained γH2AFX and TP53BP1, indicative of double-strand breaks (DSBs) and could be reversed by antioxidant treatments such as N-Acetylcysteine (NAC) or reduced glutathione ethyl ester (GSHee). The damage observed is associated with the accumulation of senescent cells, displaying a canonical senescent phenotype in both mononuclear cells and syncytia. In addition, we show signs of DNA damage and aging such as γH2AFX and CDKN2A expression in the respiratory epithelia of infected mice long after viral clearance. Altogether, these results show that HRSV triggers a DNA damage-mediated cellular senescence program probably mediated by oxidative stress. The results also suggest that this program might contribute to the physiopathology of the infection, tissue remodeling and aging, and might be associated to long-term consequences of HRSV infections.
Introduction
HRSV is an enveloped virus that belongs to the Pneumovirus genus of the Paramixoviridae family.Citation5,14 HRSV replicates in airway epithelial cells activating a variety of mediators involved in lung immune/inflammatory responses such as proinflammatory cytokines, chemokines, interferons and adhesion molecules.Citation24,26 The gene expression of these mediators is regulated by key transcription factors including nuclear factor-κB (NF-κB), the signal transducers and activators of transcription (STATs) and interferon regulatory factors (IRFs). Through its action on the signaling pathways implicated in the activation of these transcription factors, reactive oxygen species (ROS) formation can regulate the expression of the immune/inflammatory responses.Citation1,24 Excessive ROS production, however, can lead to oxidative stress and cause severe damage to cells. Thus, oxidative stress has been increasingly recognized as a contributing factor in aging and in multiple pathologies including lung inflammatory diseases.Citation38,51 HRSV infection is accompanied by the induction of ROS whose production by the pulmonary epithelial and endothelial cells is involved in the activation of transcription factors, oxidative stress and lung damage in infected cells in both animals and children.Citation9,10,24,30,31,32,37 Compelling evidence indicate that antioxidants treatments inhibit both viral HRSV replication and the activation of NF-κB, STATs and IRFs.Citation9,31,37,43 Moreover, antioxidants reduce HRSV-induced oxidative stress and clinical disease in a mouse model of infection suggesting a causal relationship between increased ROS production and lung disease.Citation10
DNA double-strand breaks (DSBs) may arise from a variety of exogenous and endogenous sources and in general by cell stress coming from any deviation from normal physiological conditions.Citation4,47,Citation55 One of the most important sources of DSBs is the generation of ROS in cells. ROS are produced primarily by the mitochondria and can induce base oxidation, abasic sites, and even both single and double-DNA strand breaks.Citation7,16,20,66 A significant number of the DNA single-stranded lesions induced by ROS are converted in DSBs, either by a direct mechanism or by the repair process itself.Citation63,72 A compelling amount of evidence has strengthened the notion that oxidative stress can trigger cellular senescence, a cellular state characterized primarily by an irreversible proliferative arrest. Besides the absence of proliferation, senescent cells display a number of morphological changes and markers that in aggregate define the senescent phenotype. These include the expression of a senescence-associated β-galactosidase activity (SA-βgal), expression of tumor suppressors and cell cycle inhibitors, secretion of diverse cytokines and often also the presence of DNA damage lesions phenotype.Citation8,15,18,61 There are a number of stimuli that induce senescence that ultimately converge in the activation of TP53 and of key cyclin-dependent kinase (CDK) inhibitors such as CDKN2A (p16), CDKN2B (p15), CDKN1A (p21) and CDKN1B (p27). These mediators trigger a proliferation arrest and the hypo-phosphorylation of the tumor suppressor RB1 that implements the cellular senescence program.Citation53 Senescence as well as apoptosis has emerged as physiological mechanisms to prevent proliferation of damaged cells. However, recent evidence has expanded its role beyond DNA damage or stress. For instance, senescence may occur during developmentCitation52,53,70 (developmental senescence) and as a physiological program in adult cells, such as normal megakaryocytes and placenta syncytiotrophoblasts,Citation73 in association with DNA damage markers, TP53, CDKN2A and CDKN1A.
HRSV induces a proliferation arrest by activating transforming growth factor β and increasing the abundance of cell-cycle regulatory molecules involved in G0/G1 phase control.Citation3,25,74 In addition, the infection courses with delayed low levels of apoptosis due to different antiapoptotic (resistance) mechanisms.Citation12,19,42,49,50 This evidence together with the fact that HRSV is able to activate the DNA damage response (DDR)Citation21 led us to investigate the role of HRSV in DNA damage and cellular senescence.
We show here the occurrence of cellular senescence during the infection of HRSV. HRSV induces the expression of DNA damage markers and proliferation arrest such as P-TP53, P-ATM, CDKN1A and γH2AFX (H2A histone family member X, phosphorylated on Ser 139) in cultured cells secondary to the production of mitochondrial ROS. The DNA damage foci contained γH2AFX and TP53BP1, indicative of DSBs and could be reversed by antioxidants treatments such as N-Acetylcysteine (NAC) or reduced glutathione ethyl ester (GSHee). The damage observed is associated with the accumulation of senescent cells, displaying all the hallmarks of the senescence phenotype in both mononuclear cells and syncytia. In addition, we show signs of DNA damage and aging such as γH2AFX and CDKN2A expression in the lungs of infected mice at different times post-infection. Altogether, these results show that oxidative stress induced by HRSV triggers a DNA damage-mediated cellular senescence program. The results also suggest that the DNA damage inflicted and the senescence program might contribute to the physiopathology of the infection, tissue remodeling and aging, and might be associated to long-term consequences of HRSV infections.
Results
HRSV induces cellular senescence in cultured cells
We observed that the infection of A549 and HEp-2 cells by HRSV induces the accumulation of senescent cells positive for SA-βgal activity. This activity could also be detected in spontaneous syncytia of both immortalized cell lines (∼20–40%), but was greatly magnified in both mononuclear cells and syncytia upon HRSV infection ( and Fig. S1a-d). The absence of significant levels of SA-βgal in the majority of the spontaneous syncytia suggests that besides the fusion process there is a specific mechanism responsible for the SA-βgal activity associated to the infection of these immortalized cells. We performed the infections 24 h after seeding cells at two densities, 5200 cells/cm2 () and 31200 cells/cm2 (Fig. S1a-b) These cellular densities correspond approximately to 18% and 33% of the typical density used in conventional infections (93750 cells/cm2) and prevent the typical SA-βgal background due to cell over confluence. Under these conditions, exposure of A549 cells to HRSV produces a drastic increase in the levels of senescence, being higher at cellular densities that favor virus dissemination (31200 cells/cm2). This effect was magnified when HEp-2 cells were used ( and Fig. S1b,c). This cell line of laryngeal origin, the cell line of choice for virus production, shows high levels of chromosomal instability and is very permissive for HRSV replication. Due to the high levels of senescence found upon HRSV infection, we used an assay consisting of a combination of SA-βgal activity and immunocytochemical staining. This technique termed here Immuno-SA-βgal allows the detection of senescent cells expressing high levels of the antigen of choice, in this case, HRSV antigens. As shown in , nearly 100% of the cells were infected (red staining) and a significant proportion of them (approximately 70%) were also positive for SA-βgal. Although to a lesser extent than in lytic infections of HEp-2 cells, significant levels of senescence could be detected in HEp-2 cells persistently infected with HRSV, i.e., cells that survived a lytic infection and were grown to obtain a culture that can be propagated indefinitely (). These cells exhibited a constant virus production for more than twenty passages and a high heterogeneity regarding the expression of viral antigens, ranging from high to undetectable levels.Citation42 Thus, the coexistence of uninfected and infected cells in these complex cultures may account for the high levels of senescence observed compared to uninfected HEp-2 cells.
Figure 1. Occurrence of cellular senescence in cells infected with HRSV. (A) Occurrence of cellular senescence during HRSV lytic infection of A549 cells seeded at 5200 cells/cm2. The cells were processed for SA-βgal assay at 48 h.p.i. (MOI = 3). Magnification: 200X, scale bar: 50 μm. (B) Quantification of SA-βgal expression in A549 cells (in total population and syncytia). (C) and (D) Similar assays in HEp-2 cells. (E) Immuno-SA-βgal, an assay that allows the detection of SA-βgal expression in massively infected HEp-2 cells stained with α-HRSV antibodies (red staining). Magnification: 200X; scale bar: 50 μm. (F) Occurrence and quantification of cellular senescence in a culture of HEp-2 cells persistently infected by HRSV. Magnification: 200X; scale bar: 50 μm. (G) Quantification of IL-6 and TNF-α in supernatants of cultured cells. Bars in the graphs represent the mean ± SD of 2–3 experiments. n = 3 replicates. (H) Analysis of apoptosis/necrosis. Eight thousand cells/well were plated in 6-well plates and were infected with HRSV the following day at MOI = 3. At 60 h.p.i., the cells were processed for flow cytometry. After treatment with the fluorescent probes (AnnexinV-AlexaFluor488™ and Sytox™) apoptotic cells show green fluorescence, dead cells show brighter green fluorescence, and live cells show little. These populations were distinguished in the FL1 channel of a FACSCalibur flow cytometer. Treatment of cells with 20 μM camptothecin (CPT) for 12 h was used as positive control for apoptosis. (I) SA-βgal assay using the fluorogenic βgalactosidase substrate C12FDG and flow cytometry (upper panel) or conventional SA-βgal (bottom panel). Eight thousand cells/well were plated in 6-well plates and were infected following day with HRSV at MOI = 3. At 60 h.p.i., the cells were incubated with Bafilomycin A1 and C12FDG and processed for flow cytometry or conventional SA-βgal assay (bottom panel) as described in materials and methods. Magnification: 200X; scale bar: 50 μm.
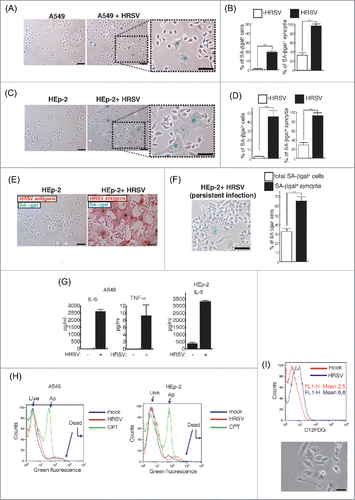
In addition to being flat and bigger than their normal counterparts, senescent cells display a senescence-associated secretory phenotype (SASP), characterized by expression of proinflammatory cytokines, growth factors and proteases. HRSV induces a rapid activation of several mediators involved in the immune/inflammatory responses. Strikingly, the broad array of proinflammatory cytokines induced by HRSV resembles the expressed SASP by canonical senescent cells.Citation5,14,60 As reported by others, we detected significantly higher levels of IL-6 and TNF-α in supernatants of infected cells when compared with mock-infected cells (). In order to know whether cellular senescence can be associated to cell survival, we assessed the levels of apoptosis and senescence after 60 h.p.i. The levels of apoptosis were determined with the Annexin V/Sytox assay. This assay allows the detection of live cells (low green fluorescence), apoptosis (moderate green fluorescence due to Annexin V binding) and necrosis (intense green fluorescence due to the Sytox green dye which is impermeable to live cells). The levels of apoptosis were similar for mock-infected and infected cells (4% approximately) and relatively low compared to the levels found when cells were treated with camptothecin, a known inducer of apoptosis (). On the other hand, the majority of cells that survived a lytic infection showed higher intensities of C12FDG fluorescence (senescence) as determined by flow cytometry analysis using this fluorogenic β-galactosidase substrate (). Cells showing FL1-H intensities over value 10 represented less than 4% of the control cultures, however, this proportion increased to 30 in the case of infected cultures. Finally, approximately 60% of cells that survived a lytic infection were senescent as determined by the conventional SA-βgal assay (). Together, these findings suggest that cellular senescence is a biological program associated to HRSV infections and might be associated to cell survival.
HRSV-induced cellular senescence is associated to DNA damage
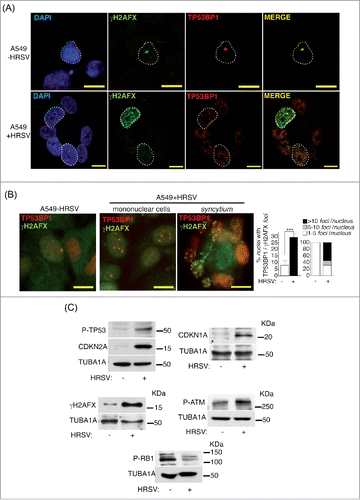
As senescence is primarily a mechanism to prevent proliferation of damaged cells, we reasoned that the senescence signs observed in virus-infected cells could be associated to DNA damage. Of the different types of DNA lesions, DSBs are the most deleterious, being potent inducers of chromosomal rearrangements such as deletions, translocations or amplifications.Citation67,75 The phosphorylated histone H2AFX (γH2AFX) and TP53BP1 are accurate markers of DNA damage.Citation2,56,62,64,65 When DSBs are detected, a signaling cascade is initiated by the phosphorylation of H2AFX by Ataxia-Telangiectasia Mutated kinase (ATM) near the break site, followed by the rapid recruitment of TP53BP1 on the chromatin surrounding the DNA lesion. TP53BP1, among other functions, acts as a molecular scaffold in damaged chromatin during non-homologous end-joining (NHEJ) repair mechanism.Citation6,56 We analyzed the expression of DNA damage (DD) foci containing γH2AFX and TP53BP1 (). First, we examined their confocal colocalization in A549 cells. As extensively reported before in other cellular systems, we corroborated their significant colocalization and observed an increase of DD foci in infected-A549 cells, in both mononuclear cells and syncytia (). Next, we performed conventional microscopy to analyze and quantify the levels of DD in the same cells and in HEp-2 cells (, Fig. S1e, f). We observed a significant increase of cells containing DD foci and cells containing numerous foci (>5/nucleus) after virus infection. This effect was exacerbated in the HEp-2 cell line (Fig. S1e, f). We further examined various markers of the DNA damage response (DDR), proliferation arrest and cellular senescence in A549 cells. As shown in , the infection of A549 cells induces the phosphorylation and activation of ATM, TP53 and H2AFX, an elevation in CDKN1A and CDKN2A expression and RB1 hypo-phosphorylation. Altogether these findings indicate that cellular senescence is a common phenomenon during HRSV infection and is associated with the presence of DNA damage.
Figure 2. HRSV induces DNA damage. (A) Analysis of confocal colocalization of the DNA damage markers γH2AFX and TP53BP1 in HRSV-infected A549 cells (48 h.p.i. MOI = 3). Magnification: 600X; scale bar: 10 μm (B) Conventional indirect immunofluorescence showing the presence of γH2AFX and TP53BP1 in mock and infected A549 cells (48 h.p.i. MOI = 3). Magnification: 600X; scale bar: 10 μm. Quantification of the DNA damage foci containing γH2AFX and TP53BP1 is shown at the right panel. (C) Detection by western-blotting of various DNA damage and proliferation arrest markers in mock and infected A549 cells (30 h.p.i. MOI = 3: panels P-TP53, CDKN2A, P-RB1, P-ATM); 48 h.p.i. MOI = 3: panels CDKN1A and γH2FAX. KDa: kilodaltons. Bars in the graphs represent the mean ± SD of 2 experiments, n = 3 replicates. DNA damage foci were counted from >150 cells for each experimental condition. Data from panel c are of a representative experiment.
Reactive oxygen species (ROS) generated during HRSV infection induce DNA damage
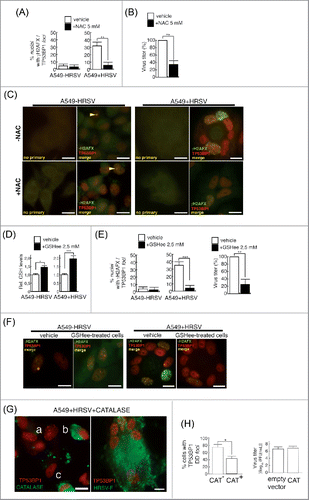
Induction of ROS by HRSV has been extensively documented.Citation9,10,24,30,31,32,37 As previously reported, we observed an elevation in the total ROS levels during the infection of A549 cells by using the widely used probe DCFH-DA Citation9,34 (). As expected, we also observed a significant increase in the intracellular amount of oxidized glutahione (GSSG) concomitantly with a decrease in reduced glutathione (GSH), upon HRSV infection as previously reported Citation32,48 (). As ROS are produced primarily by the mitochondria we further examined whether HRSV could trigger the formation of superoxide (O2.−), the main mitochondrial ROS. HRSV infection induced a drastic increase in superoxide generation, detected with the specific mitochondrial probe MitoSOX by immunofluorescence and by flow cytometry (, Fig. S2 a). Therefore, ROS triggered by HRSV have at least in part a mitochondrial origin. As ROS are able to induce DSBs and HRSV behaves as a potent stressor inducing mitochondrial superoxide, we reasoned that the DD observed could be alleviated by treatments with antioxidants as previously reported.Citation39,77 We treated cells with N-Acetylcysteine (NAC) and found a drastic reduction in the number of damaged nuclei and a concomitant moderate reduction on virus titers (). This is consistent with previous reports indicating that antioxidant treatment inhibits HRSV infectionCitation9,31,37,43 and ameliorates clinical disease and pulmonary inflammation in mice.Citation10,31 Similar results were obtained when cells were treated with reduced glutathione ethyl ester (GSHee), a permeable source of GSH (). Treatment of cells with GSHee increased the intracellular pool of GSH and induced a reduction in DD foci and virus titers. When GSHee or NAC were also included during virus adsorption, virus titers were very strongly reduced (data not shown), indicating that these antioxidants might have also an effect on virus integrity, attachment or entry into the cells. Remarkably, neither of the two treatments altered substantially the low levels of constitutive DD foci, suggesting that the antioxidants are only effective against the de novo DD foci induced by HRSV. Antioxidants treatment also reduced the occurrence of senescence in infected cultures (Fig. S2 b), and the expression or activation of DD markers such as P-TP53, γH2AFX and CDKN1A (Fig. S2 c). In addition, the partial depletion of TP53BP1 mediated by small-interfering RNAs (siRNAs) increased significatively the levels of senescence in infected cultures (Fig. S2 d,e) probably due to an additional increase of unrepaired DNA damage. In order to determine whether the oxidative stress itself was responsible of the DD observed, we transfected a plasmid encoding human catalase to HRSV infected cells and evaluated the presence of DD foci. Catalase has a preeminent role in protecting cells from oxidative damage caused by ROS.Citation36,76 Catalase calalyzes the decomposition of H2O2 to H2O and O2. H2O2 is a harmful byproduct of many normal metabolic processes including the reaction of superoxide dismutase (SOD) enzyme that converts superoxide radical into O2 and H2O. As shown in (cells labeled as “b” and “c” in the picture on the left), the overexpression of catalase reduced the occurrence of DD foci while cells with apparent signs of infection (syncytium labeled as “a” in ) showed numerous DD foci similar to those typical syncytia stained with HRSV-F glycoprotein (, picture on the right). The analysis of the DD foci and the expression of catalase in the infected/transfected population indicated that the expression of DD foci was significatively reduced in cells overexpressing catalase (, CAT+ cells, left panel). In contrast to antioxidant treatments, transfection of the plasmid encoding catalase did not alter significantly virus titers compared to cells infected and transfected with the empty vector (control culture) (, right panel). These results suggest that oxidative stress is directly responsible of the damage observed and link the occurrence of DD damage with cellular senescence. The physical presence of DNA breaks in infected cells was also directly detected by using the terminal deoxynucleotidyl transferase (TdT) enzyme,Citation77 (Fig. S2f). We also assessed the repair capability of infected cells after irradiation with γ-rays (Fig. S3 a-d). Due to the progression of the infection and the subsequent accumulation of de novo foci, the DD load was still very high compared to control cells. Analysis of the DDR at a single time suggested that HRSV does not affect significantly the early DDR signaling (Fig. S3e, f). Thus, the DD inflicted by HRSV is most likely due to oxidative stress and does not appear to be caused by a gross defect in cellular DSBs repair capacity.
Figure 3. HRSV induces mitochondrial reactive oxygen species (ROS). (A) Measurement of total ROS levels of mock and infected A549 cells (48 h.p.i. MOI = 3) using the DCFH-DA probe and fluorometry. (B) Measurement of reduced (GSH) and oxidized (GSSG) glutathione of mock and infected A549 cells (24 h.p.i. MOI = 3). (C) Evaluation of mitochondrial ROS in mock and infected A549 cells (24 h.p.i. MOI = 3) with MitoSOX and fluorescence microscopy. Positive control: cells treated with 100 μM Paraquat. Images of cells treated with Paraquat, acquired at the same exposure to that of the rest of the samples (equivalent exposure) and at a reduced exposure, are shown. Magnification: 400X; scale bar: 20 μm. (D) Assessment of mitochondrial ROS levels of mock and infected A549 cells (24 h.p.i. MOI = 3) with MitoSOX and flow cytometry. The left panel shows representative histograms. The mean intensity of MitoSOX fluorescence and the percentage of positive cells are shown at the right panel. Bars represent the mean ± SD of two experiments, ANOVA of data D: P < 0,0001; n = 3 replicates.
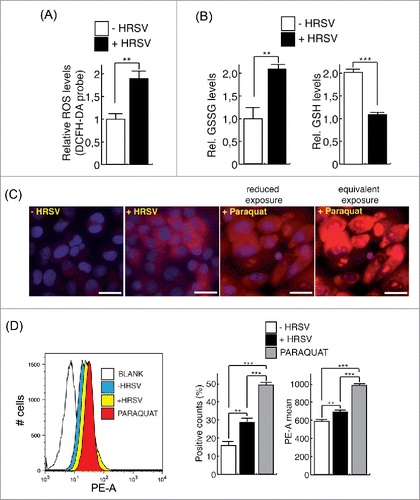
Figure 4. HRSV-induced DNA damage is reduced with antioxidant treatments (A) Quantification of DNA damage foci containing γH2AFX and TP53BP1 in mock and infected A549 cells (48 h.p.i. MOI = 3), treated or not with N-Acetylcysteine (NAC). Cells were treated with 5 mM NAC 90 min before the infection and then after virus adsorption until the end of the experiment. (B) Virus titers in supernatants of A549 infected cells (48 h.p.i. MOI = 3) treated or not with 5 mM NAC. (C) Representative images of DNA damage foci of mock and infected A549 cells (48 h.p.i. MOI = 3), treated or not with N-Acetylcysteine (NAC). Magnification: 600X; scale bar: 10 μm. Arrowheads: DD foci. (D) Increase of intracellular reduced glutathione (GSH) in mock and infected A549 cells (24 h.p.i. MOI = 3) upon treatment with 2,5 mM GSH ethyl ester (GSHee). (E) Quantification of DNA damage foci containing γH2AFX and TP53BP1 in mock and infected A549 cells (48 h.p.i. MOI = 3) treated or not with GSHee. Cells were treated with GSHee 90 min before the infection and then after virus adsorption until the end of the experiment. Virus titers in supernatants of A549 infected cells (48 h.p.i. MOI = 3) treated or not with 2,5 mM GSHee (right panel). (F) Representative images of DNA damage foci in mock and infected A549 cells (48 h.p.i. MOI = 3), treated or not with GSHee. Magnification: 600X; scale bar: 10 μm. (G) Left panel: Expression of transfected catalase (flagged-catalase, in green) and DD foci (TP53BP1 in red) in A549 cells infected with HRSV (48 h.p.i. MOI = 3) and transfected with the plasmid pCMV3-CAT-N-FLAG. Cells labeled as “b” and “c” show catalase overexpression and the absence of DD foci; syncytium labeled as “a” shows the absence of significant catalase overexpression and the presence of nuclei harboring numerous DD foci. Right panel: expression of DD foci (TP53BP1) and HRSV-F glycoprotein (green) in A549 infected cells (48 h.p.i. MOI = 3) Magnification 600X, Scale bar: 10 μM. (h) Left: panel: Quantification of DD foci in A549 cells infected with HRSV (48 h.p.i. MOI = 3) and transfected with the plasmid pCMV3-CT-N-FLAG. CAT−: cells without significant catalase overexpression, CAT+: cells overexpressing catalase. Right panel: virus titers of culture supernatants of A549 cells infected with HRSV (48 h.p.i. MOI = 3) and transfected with either empty vector as a control or catalase (CAT) plasmid. Bars represent the mean ± SD of two experimenst; n = 3 replicates. DNA damage foci were counted from >150 cells for each experimental condition.
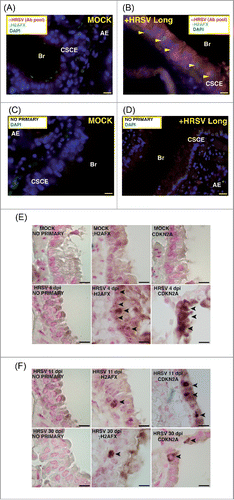
Signs of DNA damage and aging in lungs of young mice infected with HRSV
Mice were either mock infected (n = 5) or infected with HRSV (strains Long and A2, n = 6) and the lungs were analyzed by a highly sensitive multiple immunofluorescence assay to detect both γH2AFX, a marker of DD and aging,Citation44 and HRSV antigens. We found no significant differences in DD expression and viral antigens between the two strains used. As shown in and Fig. S3g, we observed signs of severely damaged DNA in 23%±2 of HRSV-infected cells, in ∼30% of the columnar epithelia of bronchioles (CSCE: ciliated simple columnar epithelium). Moderate DNA damage, 1 or 2 foci/nucleus approximately, could be also detected in ∼30% of the alveolar epithelium of infected mice (26%±5 of alveolar cells with significant reactivity to HRSV antigens showed DD foci) (data not shown). In contrast, no significant γH2AFX reactivity was detected in the lungs of mock-infected cells (). We further evaluated the expression of both γH2AFX and CDKN2A by conventional immunohistochemistry at different times post infection (4, 11 and 30 days). HRSV-infected mice normally show a viral replication that peaks at days 4–5 and becomes undetectable by day 7.Citation33 As expected, we did not observe reactivity to HRSV antigens at days 11 and 30 post infection in the lungs of infected mice (data not shown), however, we could still detect signs of DNA damage and senescence in the respiratory epitelium of some bronchioles (∼18%) at those times post infection (). These results suggest the occurrence of DNA damage and senescence long time after a primary infection.
Figure 5. Expression of markers of DNA damage and aging in lungs of mice infected by HRSV. (A) Epithelial lung tissue from mock-infected mice stained with antibodies: αHRSV (pool of antibodies against HRSV) and γH2AFX. (B) Representative image of multiple immunofluorescence labeling with αHRSV and γH2AFX antibodies of lung tissue from HRSV-infected mice stained. (C), (D) Representative images of negative controls (no primary) of the multiple immunofluorescence labeling of epithelial lung tissue. Magnifications: 600X; scale bar: 10 μm. Arrowheads signal cells showing reactivity to γH2AFX. AE: alveolar epithelium, Br: bronchiole, CSCE: ciliated simple columnar epithelium. (E) Representative images of conventional immunohistochemistry of γ-H2AFX and CDKN2A on mice lung tissue (mock-infected or infected with HRSV) at 4 days post-infection. (F) Representative images of conventional immunohistochemistry of γH2AFX and CDKN2A on mice lung tissue at 11 and 30 days post-infection. Arrowheads signal cells showing reactivity to γH2AFX and CDKN2A. Two experiments, n = 5 (mock-infected) or 6 (infected) replicates (mice) per condition.
Discussion
We show for the first time the occurrence of cellular senescence in cultured immortalized epithelial cells infected with HRSV. These infected cells show the canonical cellular senescence phenotype, including the SA-βgal activity, components of the SASP phenotype and the presence of accurate markers of DNA damage and proliferation arrest such as the phosphorylation and activation of ATM, TP53 and H2AFX, an elevation in CDKN1A and CDKN2A expression and RB1 hypo-phosphorylation. SA-βgal activity is evident at 24 h.p.i. (data not shown) but increased progressively with time and under conditions that favor virus dissemination (31200 cells/cm2), although it could be also detected in severely damaged mononuclear infected cells. Besides HRSV, fusion of cells may be also triggered by pathogenic viruses such as HIV, ERVWE1 virus, Measles, and Influenza.Citation11,28,35,46,69 This fusogenic property seems to be a mechanism to support virus dissemination and reproduction and has been associated to senescence in some cases.Citation11 In the case of HRSV and the immortalized context of the cellular systems used, it seems that cellular senescence is driven by the DNA damage induced by HRSV rather than the cell fusion process itself as signs of cellular senescence can be detected in spontaneous syncytia and in mononuclear infected cells.
As recently reported by others,Citation21 HRSV is able to activate the DDR. Here we show evidence indicating that the DNA damage inflicted by HRSV are DSBs. DSBs observed are primarily of oxidative nature, probably due to ROS generation. Several reports have previously reported that HRSV induces ROS expression by different mechanisms including nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase dependent mechanisms and inhibition of antioxidant enzymes.Citation22,24 According to the results obtained from the use of the mitochondrial superoxide indicator MitoSOX, ROS formation by HRSV has at least in part a mitochondrial origin. ROS are involved in transcription factor activation and chemokine gene expression, oxidative stress and lung damage in infected cells in both animals and children.Citation9,10,30,32,37 Antioxidants treatments inhibit viral HRSV replication and block the activation of transcription factors implicated in the regulation of immune/inflammatory responses.Citation9,31,37,43 In addition, antioxidants reduce HRSV-induced oxidative stress and clinical symptoms in mice suggesting a link between increased ROS production and lung disease.Citation10 We show that damage inflicted by HRSV can be alleviated to a great extent by antioxidants that also inhibited moderately virus production and reversed the reduced glutathione (GSH)/oxidized GSSG ratio as reported before.Citation32,48 Antioxidant treatments concomitantly reduced the levels of cellular senescence induced by HRSV and the activation of DD markers in cell cultures suggesting that the cellular senescence observed is mainly triggered by DNA damage. According to this, the ectopic expression of the enzyme catalase, that protect cells from oxidative damage from ROS, reduced the expression of DD foci in infected cells without altering virus production, in contrast to antioxidant treatments, suggesting a direct role of the oxidative stress in the damage observed. The DD inflicted does not seems to be a consequence of a gross deficiency in the cellular DD repair capabilities and the downregulation of one of the major components of DSB repair, TP53BP1, slightly increased the levels of senescence induced by HRSV probably due to increased unrepaired damage. These results suggested a direct link between DNA damage and the occurrence of senescence.
We have also shown the presence of two accurate biomarkers of DNA damage and aging in HRSV-infected lungs of young mice, γH2AFX and CDKN2A. HRSV infection was evident in some distal and terminal airways as well as in the respiratory epithelium. A significant number of γH2AFX foci were found in some infected cells lining the terminal airways. However only 1–2 foci/nucleus were observed in infected cells of the alveolar fields (data not shown). Distal airways of the mouse are lined with a simple columnar epithelium containing ciliated and secretory cells and are an important site for HRSV replication. The staining of HRSV antigens found was similar to that observed by others in the human and mouse airway columnar epithelium.Citation13,24 γH2AFX and CDKN2A expression was still evident at 11 and 30 days post infection indicating a persistence of the primary damage induced by HRSV during the acute infection. We found that CDKN2A expression pattern was diffuse nuclear and cytoplasmic. Although nuclear expression of CDKN2A is usually associated to cell-cycle control, cytoplasmic accumulation has also been associated to senescent phenotype induced by stress.Citation68
Histological studies suggest that non-ciliated secretory cells give rise to ciliated cells and function as a long-term self-renewing stem cell population in the intrapulmonary airways.Citation59 In addition, it has been reported that the adult mouse lung contains a minor population of multipotent epithelial stem cells with the capacity of self-renewal and to give rise to all epithelial progenitors that reside in discrete microenviromental niches along the proximal-distal axis.Citation45 It is tempting to speculate that such stem cells could be targets of HRSV. Indeed, it has been recently reported that HRSV can infect proliferating airway basal cells and alter cellular composition of the epithelium contributing to airway epithelial remodeling.Citation58 Thus, the DNA damage inflicted and the occurrence of senescence might influence events long time after the virus has cleared from the lung by mechanisms based on a kind of “hit-and-run” phenomenon that has been proposed to explain how transient infections can cause long term airway disease.Citation29
Our findings might have substantial implications for the pathophysiology of HRSV, the leading cause of bronchiolitis and pneumonia in infants and young children.Citation27,54 During the acute infection, the SASP might contribute to alert the immune system about the damage inflicted. The SASP components would mediate this process, facilitating the attraction of immune cells to the infection sites and the further clearance of infected cells.
Finally, the SASP and cellular senescence may serve as therapeutic targets.Citation53,57,71 Although it is too premature to determine whether cellular senescence would be positive or detrimental for the pathological consequences of HRSV infection, current evidence indicates that both prosenescent and antisenescent therapeutic approaches can be useful depending on the context. To confirm this hypothesis further investigations in human primary cells and in more permissive animal models supporting HRSV infections are needed.
Methods
Cell culture, viruses, mice and transfections
HEp-2 and A549 cells (ATCC) were maintained in DMEM medium supplemented with 10% FBS (Biowest), 2 mM glutamine and 100 U/ml of penicillin and streptomycin (Lonza). The Long and A2 strains of HRSV were propagated in HEp-2 cells in medium supplemented with 2% FBS, glutamine and antibiotics. Viruses were purified and titrated in HEp-2 cells as previously described.Citation41 All experiments were performed with the Long strain except for the in vivo experiments in which the A2 strain was also used. Two-month-old female mice (BALB/c strain) were intranasally infected with 3,3×107 pfus of purified virus. At 4, 11 and 30 days post-infection, mice were sacrified by cervical dislocation and the lungs were processed for immunohistochemistry. All experiments were performed following the regulations of the Instituto Carlos III for animal care and handling. HEp-2 persistently infected by HRSV (L39) were described elsewhere.Citation42 N-Acetylcysteine (NAC, Sigma) and reduced glutathione reduced ethyl ester (GSHee, Sigma) were prepared as indicated by the manufacturers and used at 5 mM and 2,5 mM, respectively. Tansfections of siRNAs were performed with Lipofectamine RNAimax (cat.#13778, Invitrogen) following manufacturers instructions. Briefly, cells were transfected with siRNAs (control or TP53BP1 duplexes, sc-37007 and sc-37455, respectively, Santa Cruz Biotechnology) and the following day infected at MOI 3 with HRSV. After 48 h.p.i., the cells were processed for western-blotting and SA-βgal assays. Transitory transfections of the plasmid pCMV3-CT-N-FLAG (HG12084-NF, Sino Biological Inc.) expressing human catalase or empty vector (pCMV) were performed with Lipofectamine 2000 (Invitrogen, cat. 11668) in 8-well chamber slides following manufacturers instructions. Cells were transfected and the following day infected at MOI 3 with HRSV. After 48 h.p.i., culture supernatants were collected for virus titration and the cells were processed for immunofluorescence.
Immunofluorescence
For immunofluorescence, cells were seeded in 8-well chambers (Thermo Fisher Scientific) at a density of 50000 cells/well. The following day the cells were infected or treated as indicated in the corresponding experiments. Immunofluorescence was performed as previously described.Citation77 Basically, cells were fixed in 2% PFA in PBS for 10 min at RT and permeabilized with 0.1% Triton X-100 and 0.1% sodium citrate (5 min at RT). Preparations were then washed with PBS and washing solution (PBS/0.25% BSA/0.1% Tween 20), blocked for 30 min with blocking solution (washing solution, 2.5% BSA, and 5% normal serum), and incubated overnight with antibodies against γH2AFX (1:1000, cat.# 05–636 Millipore), TP53BP1 (1:1000, cat.# NB-100–304, Novus Biologicals), antibody 47F (1:50) directed against the F glycoprotein of HRSV and anti-Flag (1:500, cat.# F1804, Sigma-Aldrich). Preparations were then washed with washing solution and incubated with secondary antibodies (AF488, AF546 or AF647, Life Technologies) for 1 h at RT. Nuclei were counterstained with DAPI, and samples were mounted with ProLong Diamond (Life Technologies). Cell images were captured with a fluorescence microscopy (Zeiss Axio) equipped with a camera (Axiocam MRm) and AxioVision software. DNA damage foci were counted from >150 cells for each experimental condition. Analysis of colocalization was performed with a confocal microscope (LEICA DMI 600; 63X/1.4 len), equipped with the following laser lines: Argon (five spectral lines 488 to 514 nm), HeNe (633 nm), diode violet (405 nm) and diode solid state (561 nm). Images were obtained with software LAS AF (Leica).
Protein analysis
Cell monolayers were washed with ice-cold PBS and lysed in triple-detergent lysis buffer [50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.02% sodium azide, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, 100 μg/ml PMSF, 2 μg/ml pepstatin, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and phosphatase inhibitors cocktail 2 and 3 (Sigma-Aldrich)]. SDS-PAGE and immunoblotting were performed under standard conditions. Basically, samples in Laemmli buffer (30 μg/lane) were separated through 8% or 12% gels and transferred to nitrocellulose membranes for 90 min at RT in the presence of 20% methanol and 0,1% SDS. Membranes were blocked with 3% BSA in PBS-Tween 0,05% (PBST-BSA) and incubated ON at 4°C with specific antibodies diluted 1:1000 in PBST-BSA. Antibodies used were: γH2AFX (05–636, Millipore), P-CHEK2 (AF1626, R&D Systems), P-RB1 (sc-16670-R, Santa Cruz Biotech.), P-TP53 (9286S, Cell Signaling), P-ATM (05–740, Millipore), CDKN1A (ab7960, Abcam), CDKN2A (04–239, Millipore), TP53BP1 (cat.# NB-100–304, Novus Biologicals) and TUBA1A (used at 1:2000, clone DM1A, Sigma-Aldrich).
Senescence-associated β-galactosidase assays (SA-βgal) and Immuno-SA-βgal
Conventional SA-βgal assays were performed as previously described by Dimri et al.Citation18 SA-βgal detection by flow cytometry using Bafilomycin A1 (cat.#B1793, Sigma-Aldrich) and the fluorogenic βgalactosidase substrate C12FDG (cat.# 11590276, Fisher Scientific) was performed as described by Debacq-Chainiaux et al.Citation17 Immuno-SA-βgal is an assay consisting of a combination of immunocytochemistry and SA-βgal assays. Cells growing in 6-well plates (MW6) were washed twice with PBS and fixed for 3 minutes with 2% formaldehyde and 0,2% glutaraldehyde. Cells were permeabilized for 5 min at RT with 0.1% Triton X-100 and 0.1% sodium citrate, washed with PBS and blocked with BSA 1% in PBS (30 min at RT). Cells were then incubated for 45 min at RT with a pool of antibodies directed against HRSV [021/1G, 021/2G, 47F, 67P].Citation23,40 The plates were washed twice in a bath of tap water and incubated for 45 min at RT with α-mouse-HRP secondary (ab97040, Abcam) 1:1000 in PBS-BSA 1%. The plates were washed with tap water before adding the peroxidase substrate in ELISA buffer [Citric acid 25 mM/Na2HPO4 50 mM pH 5, 0,033 mg/ml AEC substrate (A6926-100TAB, Sigma), 2 μl/ml (30% H2O2)]. The plates were maintained in the dark until the appearance of signal (5–10 min). After that, the substrate was removed, the plates were washed twice with PBS and the SA-β-gal reaction mixture was added. The plates were incubated in the dark at 37° for 24 hours. Micrographs were taken in a microscope TS100F (Nikon) equipped with a digital camera DS-L1 (Nikon).
ROS analyses and quantification of GSSG (oxidized glutathione) and GSH (reduced glutathione)
Cellular ROS levels were assessed with 20 μM H2DCFDA (2′,7′ dichlorodihydrofluorescein diacetate, Sigma-Aldrich) and a microplate fluorometer Synergy (Biotek) as previously described.Citation77 For the determination of mitochondrial ROS (superoxide) we used MitoSOX Red (Life Technologies), fluorescence microscopy and flow citometry. Treatments of cells were carried-out in triplicates in 8-well chambers (for microscopy) and in MW6 plates (for flow cytometry). Positive controls consisted of cells treated with 5 μM MitoSOX for 15 min, then with 100 μM Paraquat for 30 min followed by an additional 30 min treatment with 5 μM MitoSOX. The rest of the samples were treated with 5 μM MitoSOX for 30 min. Blank cells consisted of cells treated with vehicle. The cells growing in 8-well chambers were fixed with PFA 2% in PBS for 10 min, washed and incubated with DAPI for 15 minutes. Then, the cells were washed with PBS, mounted with ProLong Diamond (Life Technologies) and examined under fluorescence microscopy. For flow cytometry, the cells were trypsinized and counted. The same number of cells for each condition was fixed with PFA, washed and transferred to FACS tubes. The measurements were carried out using FASCanto (BD Bioscience) cytometer. MitoSOX was excited by a laser at 488 nm and the data collected at the FSC, SSC, 585/42 nm bandpass filter. Cell debris, represented by distinct low forward and side scatter, were gated out for analysis. 50000 gated events for each condition were analyzed with the FACSCanto and Flow Jo software. The mean intensity of MitoSOX fluorescence and the percentage of positive cells were calculated. For the determination of GSSG and GSH we used the EnzyChrom™ GSH/GSSG Assay kit (Bioassay Systems) and followed manufacturer's instructions.
Apoptosis/necrosis by flow cytometry
This assay was performed by using the single channel dead cells apoptosis kit with Annexin V-Alexa-fluor488™ and Sytox™ green dyes from Fisher Scientific (cat.# V13240). Eight thousand cells per well were plated in MW6 and were infected with HRSV at MOI 3 the following day. Sixty h.p.i., attached cells were processed for flow cytometry as indicated by the manufacturers. Treatment of cells with 20 μM camptothecin (CPT) for 12 h (cat.#C9911, Sigma-Aldrich) was used as positive control for apoptosis. After treatment with both probes, apoptotic cells show green fluorescence, dead cells show brighter green fluorescence, and live cells show little. These populations were distinguished in the FL1 channel of a FACSCalibur flow cytometer (Becton Dickinson). Twenty thousand events for each condition were analyzed with the FACSCalibur and Flow Jo software.
Immunohistochemistry
Mice were sacrificed at 4, 11 and 30 days post-infection by cervical dislocation and lung pieces (5 × 5 mm, approximately) were dissected, fixed in 4% buffered formalin and embedded in paraffin wax. Multiple antigen labeling was performed following the double-immunofluorescent labeling protocol and reagents provided by Vector Laboratories (cat. # A-2011, A-2016, SP-2001, PK-6102). Sequential incubations of the antibodies were made overnight for the first antibody (α-HRSV: pool of Abs 021/1G, 021/2G, 47F, 67P, 1:100) and 3 h at RT for the second antibody (γH2AFX, 1:400). Antigen retrieval was performed with citrate buffer (pH 6) using a Microwave Tender Cooker (Nordic Ware) and a 700 W microwave (15 min 700W/15 min 350W). Endogenous peroxidase activity was inhibited with 0.3% H2O2 in methanol (25 min). Nuclei staining, mounting, and microscopy acquisition were performed as for the indirect immunofluorescence. Images of were processed by deconvolution in the green channel with Huygens Essential (Scientific Volume Imaging, Hilversum, the Netherlands) based on the classicmaximum likelihood estimation method. Conventional immunohistochemistry was performed following the labeling protocol and reagents provided by Vector Laboratories (PK-6101, PK-6102, SP-2001). Antibodies used were γH2AFX (1:200 cat.# 05–636 Millipore) and CDKN2A (1:200, cat.# 04–239, Millipore). Counterstaining with nuclear fast, mounting and images acquisition were performed as previously described.Citation77
Immunoassay
The determination of the concentration of human IL-6 and TNF-α from cell culture supernatatants was assessed by the ProcartaPlex™ multiplex Immunoassay kit -Magnetic beads (eBioscence) based on the LuminexR technology. Procarta PlexTM Analyst 1.0 software was used for analysis.
Irradiation of cells with γ-ray
Cells growing in 8-well chambers and MW6 plates were irradiated with 3 Gy (1.2 Gy/min) in a shelf-shielded J. L. Shepherd Mark I irradiator with a Cesium-137 source. After irradiation cells were processed for immunofluorescence or immunoblotting at the corresponding times.
DNA breaks detection
To detect DNA breaks, we performed an assay based in the incorporation of dUTP by Terminal deoxynucleoitidyl Transferase enzyme (TdT) and posterior detection by immunocytochemistry as previously described.Citation77
Statistical analysis
Statistical significance of data was determined by applying a two-tailed Student's t test or analysis of variance followed by the Newman–Keuls or Bonferroni post-tests for experiments with more than two experimental groups. P <0.05 is considered significant. Significance of analysis of variance post-test or the Student's t test is indicated in the figures as *(P < 0.05), **(P <0.01) and ***(P < 0.001). Statistics were calculated with the Prism 5 software (GraphPad Software). The results presented in the figures are means ±SD. Experiments were repeated at least two times.
Ethics statement
All experiments with animals were approved by the Commitee of Bioethics and Animal Welfare of the Instituto de Salud Carlos III (file reference: PA 31). Protocols used followed the guidelines for animal protection reported by the Spanish national law RD 53/2013.
Abbreviations
| CPT | = | Camptothecin |
| DD | = | DNA damage |
| DDR | = | DNA damage response |
| DSB | = | double-strand breaks |
| GSH | = | reduced glutathione |
| GSHee | = | reduced glutathione ethyl ester |
| GSSG | = | oxidized glutathione |
| h.p.i | = | hours post-infection |
| MOI | = | multiplicity of infection |
| MW6 | = | 6-well plates |
| NAC | = | N-Acetylcysteine |
| ROS | = | reactive oxygen species |
| siRNA | = | small interfering RNAs |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
IM performed and designed experiments. V G-C performed and designed experiments. TG, A G-G and DG performed experiments, AA designed experiments and wrote paper. AZ performed experiments, designed experiments, wrote paper and conceived the project.
KVIR_A_1144001.pdf
Download PDF (9.2 MB)Acknowledgments
We thank the core facilities of the Instituto de Investigaciones Biomédicas de Madrid and of the Centro Nacional de Microbiología for technical help.
Funding
This work was supported by Grants MPY-1038/14 to Alberto Zambrano, PI 11/00590 to Isidoro Martínez and RD12/0036/0030 to Ana Aranda from FIS (Instituto de Salud Carlos III) and Grant BFU2011-28958, from Ministerio de Economía y Competitividad to Ana Aranda. The authors have no conflicting financial interests.
References
- Allen RG, Tresini M. Oxidative stress and gene regulation. Free Radical Biol Med 2000; 28:463-99; http://dx.doi.org/10.1016/S0891-5849(99)00242-7
- Anderson L, Henderson C, Adachi Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol 2001; 21:1719-29; PMID:11238909; http://dx.doi.org/10.1128/MCB.21.5.1719-1729.2001
- Bian T, Gibbs JD, Orvell C, Imani F. Respiratory syncytial virus matrix protein induces lung epithelial cell cycle arrest through a p53 dependent pathway. PloS One 2012; 7:e38052; PMID:22662266; http://dx.doi.org/10.1371/journal.pone.0038052
- Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer 2008; 8:957-67; PMID:19005492; http://dx.doi.org/10.1038/nrc2523
- Borchers AT, Chang C, Gershwin ME, Gershwin LJ. Respiratory syncytial virus–a comprehensive review. Clin Rev Allergy Immunol 2013; 45:331-79; PMID:23575961; http://dx.doi.org/10.1007/s12016-013-8368-9
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243-54; PMID:20362325; http://dx.doi.org/10.1016/j.cell.2010.03.012
- Cadet J, Douki T, Ravanat JL. Oxidatively generated base damage to cellular DNA. Free Radical Biol Med 2010; 49:9-21; http://dx.doi.org/10.1016/j.freeradbiomed.2010.03.025
- Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nature Rev Mol Cell Biol 2007; 8:729-40; http://dx.doi.org/10.1038/nrm2233
- Casola A, Burger N, Liu T, Jamaluddin M, Brasier AR, Garofalo RP. Oxidant tone regulates RANTES gene expression in airway epithelial cells infected with respiratory syncytial virus. Role in viral-induced interferon regulatory factor activation. J Biol Chem 2001; 276:19715-22; PMID:11259439; http://dx.doi.org/10.1074/jbc.M101526200
- Castro SM, Guerrero-Plata A, Suarez-Real G, Adegboyega PA, Colasurdo GN, Khan AM, Garofalo RP, Casola A. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am J Respiratory Critical Care Med 2006; 174:1361-9; http://dx.doi.org/10.1164/rccm.200603-319OC
- Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, Krizhanovsky V. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev 2013; 27:2356-66; PMID:24186980; http://dx.doi.org/10.1101/gad.227512.113
- Coleman CM, Plant K, Newton S, Hobson L, Whyte MK, Everard ML. The anti-apoptotic effect of respiratory syncytial virus on human peripheral blood neutrophils is mediated by a monocyte derived soluble factor. Open Virol J 2011; 5:114-23; PMID:22046209; http://dx.doi.org/10.2174/1874357901105010114
- Collins PL, Graham BS. Viral and host factors in human respiratory syncytial virus pathogenesis. J Virol 2008; 82:2040-55; PMID:17928346; http://dx.doi.org/10.1128/JVI.01625-07
- Collins PL, Karron RA. Respiratory Syncytial Virus and Metapneumovirus. Philadelphia: Lippincott Williams & Wilkins. 2013
- d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 2008; 8:512-22; PMID:18574463; http://dx.doi.org/10.1038/nrc2440
- De Zio D, Bordi M, Cecconi F. Oxidative DNA damage in neurons: implication of ku in neuronal homeostasis and survival. Int J Cell Biol 2012; 2012:752420; PMID:22737170; http://dx.doi.org/10.1155/2012/752420
- Debacq-Chainix F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated β-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 2009; 4:1798-806; PMID:20010931; http://dx.doi.org/10.1038/nprot.2009.191
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995; 92:9363-7; PMID:7568133; http://dx.doi.org/10.1073/pnas.92.20.9363
- Domachowske JB, Bonville CA, Mortelliti AJ, Colella CB, Kim U, Rosenberg HF. Respiratory syncytial virus infection induces expression of the anti-apoptosis gene IEX-1L in human respiratory epithelial cells. J Infect Dis 2000; 181:824-30; PMID:10720500; http://dx.doi.org/10.1086/315319
- Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutation Res 2004; 567:1-61; PMID:15341901; http://dx.doi.org/10.1016/j.mrrev.2003.11.001
- Fang L, Choudhary S, Tian B, Boldogh I, Yang C, Ivanciuc T, Ma Y, Garofalo RP, Brasier AR. Ataxia telangiectasia mutated kinase mediates NF-kappaB serine 276 phosphorylation and interferon expression via the IRF7-RIG-I amplification loop in paramyxovirus infection. J Virol 2015; 89:2628-42; PMID:25520509; http://dx.doi.org/10.1128/JVI.02458-14
- Fink K, Duval A, Martel A, Soucy-Flkner A, Grandvx N. Dual role of NOX2 in respiratory syncytial virus- and sendai virus-induced activation of NF-kappaB in airway epithelial cells. J Immunol 2008; 180:6911-22; PMID:18453612; http://dx.doi.org/10.4049/jimmunol.180.10.6911
- Garcia-Barreno B, Palomo C, Penas C, Delgado T, Perez-Brena P, Melero JA. Marked differences in the antigenic structure of human respiratory syncytial virus F and G glycoproteins. J Virol 1989; 63:925-32; PMID:2463385
- Garofalo RP, Kolli D, Casola A. Respiratory syncytial virus infection: mechanisms of redox control and novel therapeutic opportunities. Antioxidants Redox Signal 2013; 18:186-217; http://dx.doi.org/10.1089/ars.2011.4307
- Gibbs JD, Ornoff DM, Igo HA, Zeng JY, Imani F. Cell cycle arrest by transforming growth factor beta1 enhances replication of respiratory syncytial virus in lung epithelial cells. J Virol 2009; 83:12424-31; PMID:19759128; http://dx.doi.org/10.1128/JVI.00806-09
- Haeberle HA, Takizawa R, Casola A, Brasier AR, Dieterich HJ, Van Rooijen N, Gatalica Z, Garofalo RP. Respiratory syncytial virus-induced activation of nuclear factor-kappaB in the lung involves alveolar macrophages and toll-like receptor 4-dependent pathways. J Infect Dis 2002; 186:1199-206; PMID:12402188; http://dx.doi.org/10.1086/344644
- Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, et al. The burden of respiratory syncytial virus infection in young children. N Eng J Med 2009; 360:588-98; http://dx.doi.org/10.1056/NEJMoa0804877
- Harrison SC. Viral membrane fusion. Nat Struct Mol Biol 2008; 15:690-8; PMID:18596815; http://dx.doi.org/10.1038/nsmb.1456
- Holtzman MJ, Shornick LP, Grayson MH, Kim EY, Tyner JW, Patel AC, Agapov E, Zhang Y. “Hit-and-run” effects of paramyxoviruses as a basis for chronic respiratory disease. Pediatric Infect Dis J 2004; 23:S235-245; http://dx.doi.org/10.1097/01.inf.0000144674.24802.c1
- Hosakote YM, Jantzi PD, Esham DL, Spratt H, Kurosky A, Casola A, Garofalo RP. Viral-mediated inhibition of antioxidant enzymes contributes to the pathogenesis of severe respiratory syncytial virus bronchiolitis. Am J Respiratory Critical Care Med 2011; 183:1550-60; http://dx.doi.org/10.1164/rccm.201010-1755OC
- Hosakote YM, Komaravelli N, Mautemps N, Liu T, Garofalo RP, Casola A. Antioxidant mimetics modulate oxidative stress and cellular signaling in airway epithelial cells infected with respiratory syncytial virus. Am J Physiol Lung Cell Mol Physiol 2012; 303:L991-1000; PMID:23023968; http://dx.doi.org/10.1152/ajplung.00192.2012
- Hosakote YM, Liu T, Castro SM, Garofalo RP, Casola A. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am J Resp Cell Mol Biol 2009; 41:348-57; http://dx.doi.org/10.1165/rcmb.2008-0330OC
- Jafri HS, Chavez-Bueno S, Mejias A, Gomez AM, Rios AM, Nassi SS, Yusuf M, Kapur P, Hardy RD, Hatfield J, et al. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long-term airway hyperresponsiveness in mice. J Infect Dis 2004; 189:1856-65; PMID:15122522; http://dx.doi.org/10.1086/386372
- Jamaluddin M, Tian B, Boldogh I, Garofalo RP, Brasier AR. Respiratory syncytial virus infection induces a reactive oxygen species-MSK1-phospho-Ser-276 RelA pathway required for cytokine expression. J Virol 2009; 83:10605-15; PMID:19706715; http://dx.doi.org/10.1128/JVI.01090-09
- Jardetzky TS, Lamb RA. Activation of paramyxovirus membrane fusion and virus entry. Curr Opin Virol 2014; 5:24-33; PMID:24530984; http://dx.doi.org/10.1016/j.coviro.2014.01.005
- Lindau-Shepard BA, Shaffer JB. Expression of human catalase in acatalasemic murine SV-B2 cells confers protection from oxidative damage. Free Radical Biol Med 1993; 15:581-8; http://dx.doi.org/10.1016/0891-5849(93)90160-V
- Liu T, Castro S, Brasier AR, Jamaluddin M, Garofalo RP, Casola A. Reactive oxygen species mediate virus-induced STAT activation: role of tyrosine phosphatases. J Biol Chem 2004; 279:2461-9; PMID:14578356; http://dx.doi.org/10.1074/jbc.M307251200
- MacNee W. Oxidative stress and lung inflammation in airways disease. Eur J Pharmacol 2001; 429:195-207; PMID:11698041; http://dx.doi.org/10.1016/S0014-2999(01)01320-6
- Mansour HH, Hafez HF, Fahmy NM, Hanafi N. Protective effect of N-acetylcysteine against radiation induced DNA damage and hepatic toxicity in rats. Biochem Pharmacol 2008; 75:773-80; PMID:18028880; http://dx.doi.org/10.1016/j.bcp.2007.09.018
- Martinez I, Dopazo J, Melero JA. Antigenic structure of the human respiratory syncytial virus G glycoprotein and relevance of hypermutation events for the generation of antigenic variants. J General Virol 1997; 78 (Pt 10):2419-29; http://dx.doi.org/10.1099/0022-1317-78-10-2419
- Martinez I, Lombardia L, Garcia-Barreno B, Dominguez O, Melero JA. Distinct gene subsets are induced at different time points after human respiratory syncytial virus infection of A549 cells. J General Virol 2007; 88:570-81; http://dx.doi.org/10.1099/vir.0.82187-0
- Martinez I, Lombardia L, Herranz C, Garcia-Barreno B, Dominguez O, Melero JA. Cultures of HEp-2 cells persistently infected by human respiratory syncytial virus differ in chemokine expression and resistance to apoptosis as compared to lytic infections of the same cell type. Virology 2009; 388:31-41; PMID:19345972; http://dx.doi.org/10.1016/j.virol.2009.03.008
- Mata M, Martinez I, Melero JA, Tenor H, Cortijo J. Roflumilast inhibits respiratory syncytial virus infection in human differentiated bronchial epithelial cells. PloS One 2013; 8:e69670; PMID:23936072; http://dx.doi.org/10.1371/journal.pone.0069670
- Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Vina J, Blasco MA, Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature 2007; 448:375-9; PMID:17637672; http://dx.doi.org/10.1038/nature05949
- McQualter JL, Yuen K, Williams B, Bertoncello I. Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc Natl Acad Sci U S A 2010; 107:1414-9; PMID:20080639; http://dx.doi.org/10.1073/pnas.0909207107
- Melikyan GB. HIV entry: a game of hide-and-fuse? Curr Opin Virol 2014; 4:1-7; PMID:24525288; http://dx.doi.org/10.1016/j.coviro.2013.09.004
- Mladenov E, Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutation Res 2011; 711:61-72; PMID:21329706; http://dx.doi.org/10.1016/j.mrfmmm.2011.02.005
- Mochizuki H, Todokoro M, Arakawa H. RS virus-induced inflammation and the intracellular glutathione redox state in cultured human airway epithelial cells. Inflammation 2009; 32:252-64; PMID:19548075; http://dx.doi.org/10.1007/s10753-009-9128-0
- Monick MM, Cameron K, Powers LS, Butler NS, McCoy D, Mallampalli RK, Hunninghake GW. Sphingosine kinase mediates activation of extracellular signal-related kinase and Akt by respiratory syncytial virus. Am J Resp Cell Mol Biol 2004; 30:844-52; http://dx.doi.org/10.1165/rcmb.2003-0424OC
- Monick MM, Cameron K, Staber J, Powers LS, Yarovinsky TO, Koland JG, Hunninghake GW. Activation of the epidermal growth factor receptor by respiratory syncytial virus results in increased inflammation and delayed apoptosis. J Biol Chem 2005; 280:2147-58; PMID:15542601; http://dx.doi.org/10.1074/jbc.M408745200
- Morcillo EJ, Estrela J, Cortijo J. Oxidative stress and pulmonary inflammation: pharmacological intervention with antioxidants. Pharmacol Res 1999; 40:393-404; PMID:10527653; http://dx.doi.org/10.1006/phrs.1999.0549
- Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, et al. Programmed cell senescence during mammalian embryonic development. Cell 2013; 155:1104-18; PMID:24238962; http://dx.doi.org/10.1016/j.cell.2013.10.019
- Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014; 15:482-96; PMID:24954210; http://dx.doi.org/10.1038/nrm3823
- Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O'Brien KL, Roca A, Wright PF, Bruce N, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 2010; 375:1545-55; PMID:20399493; http://dx.doi.org/10.1016/S0140-6736(10)60206-1
- O'Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat Rev Genetics 2006; 7:45-54; PMID:16369571; http://dx.doi.org/10.1038/nrg1746
- Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 2014; 15:7-18; PMID:24326623; http://dx.doi.org/10.1038/nrm3719
- Perez-Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer 2014; 14:547-58; PMID:25030953; http://dx.doi.org/10.1038/nrc3773
- Persson BD, Jaffe AB, Fearns R, Danahay H. Respiratory syncytial virus can infect basal cells and alter human airway epithelial differentiation. PloS One 2014; 9:e102368; PMID:25033192; http://dx.doi.org/10.1371/journal.pone.0102368
- Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, Wang F, Hogan BL. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 2009; 4:525-34; PMID:19497281; http://dx.doi.org/10.1016/j.stem.2009.04.002
- Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192:547-56; PMID:21321098; http://dx.doi.org/10.1083/jcb.201009094
- Rodier F, Campisi J, Bhaumik D. Two faces of p53: aging and tumor suppression. Nucleic Acids Res 2007; 35:7475-84; PMID:17942417; http://dx.doi.org/10.1093/nar/gkm744
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273:5858-68; PMID:9488723; http://dx.doi.org/10.1074/jbc.273.10.5858
- Roth DB, Wilson JH. Illegitimate recombination in mammalian cells. In: Kucherlapati R, Smith G. (eds) Genetic Recombination. ASM Press, Washington, DC 1988, pp. 621-53
- Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 2000; 151:1381-90; PMID:11134068; http://dx.doi.org/10.1083/jcb.151.7.1381
- Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol 2004; 6:168-70; PMID:14755273; http://dx.doi.org/10.1038/ncb1095
- Sedelnikova OA, Redon CE, Dickey JS, Nakamura AJ, Georgakilas AG, Bonner WM. Role of oxidatively induced DNA lesions in human pathogenesis. Mutation Res 2010; 704:152-9; PMID:20060490; http://dx.doi.org/10.1016/j.mrrev.2009.12.005
- Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res 2008; 18:134-47; PMID:18157161; http://dx.doi.org/10.1038/cr.2007.111
- Spallarossa P, Altieri P, Barisione C, Passalacqua M, Aloi C, Fugazza G, Frassoni F, Podesta M, Canepa M, Ghigliotti G, et al. p38 MAPK and JNK antagonistically control senescence and cytoplasmic p16INK4A expression in doxorubicin-treated endothelial progenitor cells. PloS One 2010; 5:e15583; PMID:21187925; http://dx.doi.org/10.1371/journal.pone.0015583
- Stegmann T. Membrane fusion mechanisms: the influenza hemagglutinin paradigm and its implications for intracellular fusion. Traffic 2000; 1:598-604; PMID:11208147; http://dx.doi.org/10.1034/j.1600-0854.2000.010803.x
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013; 155:1119-30; PMID:24238961; http://dx.doi.org/10.1016/j.cell.2013.10.041
- Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013; 123:966-72; PMID:23454759; http://dx.doi.org/10.1172/JCI64098
- Thompson L, Limoli C. Origin, Recognition, signaling and repair of DNA double-strand breaks in mammalian cells. In: Caldecott KW, editor. Eukaryotic DNA Damage Surveillance and Repair. Georgetown, Texas: Landes Bioscience; 2004; 2004: p 107-45.
- Ullah Z, Lee CY, Lilly MA, DePamphilis ML. Developmentally programmed endoreduplication in animals. Cell Cycle 2009; 8:1501-9; PMID:19372757; http://dx.doi.org/10.4161/cc.8.10.8325
- Wu W, Munday DC, Howell G, Platt G, Barr JN, Hiscox JA. Characterization of the interaction between human respiratory syncytial virus and the cell cycle in continuous cell culture and primary human airway epithelial cells. J Virol 2011; 85:10300-9; PMID:21795354; http://dx.doi.org/10.1128/JVI.05164-11
- Wyman C, Kanaar R. DNA double-strand break repair: all's well that ends well. Annual Rev Genetics 2006; 40:363-83; http://dx.doi.org/10.1146/annurev.genet.40.110405.090451
- Ye G, Metreveli NS, Donthi RV, Xia S, Xu M, Carlson EC, Epstein PN. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 2004; 53:1336-43; PMID:15111504; http://dx.doi.org/10.2337/diabetes.53.5.1336
- Zambrano A, Garcia-Carpizo V, Gallardo ME, Villamuera R, Gomez-Ferreria MA, Pascual A, Buisine N, Sachs LM, Garesse R, et al. The thyroid hormone receptor β induces DNA damage and premature senescence. J Cell Biol 2014; 204:129-46; PMID:24395638; http://dx.doi.org/10.1083/jcb.201305084