
Introduction
Human respiratory syncytial virus (hRSV) is the major cause of respiratory tract infection in infants and young children, and it is estimated that close to 100% of children have faced a hRSV infection when reaching the age of 2. In 0.5 to 2% of the cases, hospitalization is required. Complications sometimes severe and life threatening can occur, particularly in groups at risk such as infants born prematurely or with a pre-existing health condition. Thus, hRSV is an important health problem worldwide with an economic impact estimated higher than influenza.Citation1-5
In the present issue of Virulence, Martinez et al. describe for the first time that hRSV is able to trigger the premature senescence of its target cells. Formation of DNA damage, including DNA double-strand breaks was observed and linked to the entry of cells into senescence. Moreover, a marked increase in reactive oxygen species (ROS) production is recorded in infected cells and constitutes the trigger for DNA damage formation and senescence induction.Citation6
Replicative senescence and stress-induced premature senescence
The so-called “Hayflick limit” indicates that normal somatic cells can achieve a finite number of divisions, e.g. typically 50 for human fibroblasts, before undergoing an irreversible growth arrest. Replicative senescence is not synonym of cell death. Indeed, senescent cells remain alive and metabolically active for a long period of time.Citation7-11 Senescent cells display different characteristics such as an enlarged and flattened morphology (“egg on a plate”), expression of the senescence-associated β-galactosidase activity (SA-βgal activity), accumulation of lipofuscin, presence of DNA damage, multiple changes in gene expression, presence of heterochromatin foci and secretion of pro-inflammatory cytokines and other factors.Citation7-11 Replicative senescence is also referred to as telomere-initiated senescence. Due to the end-replication problem, telomeres undergo a shortening at each cell cycle S phase, establishing a counting mechanism or mitotic clock.Citation9-14 Telomeres end in a specialized circular structure stabilized by the shelterin complex. While, in long telomeres, the extremity of the chromosome is fully masked inside this structure, a critically short telomere becomes dysfuntional and generates a robust DNA damage response.Citation9-14
Normal cells can also reach in a matter of days an irreversible post-mitotic state when exposed to different stress. This process has been called stress-induced premature senescence (SIPS). Remarkably, replicative senescence and SIPS share many features, including morphological changes and expression of the SA-βgal activity.Citation15-18 Cells enter SIPS following a variety a detrimental stimuli ().Citation19,20 Moreover, SIPS is independent of telomere shortening. Cells that have escaped replicative senescence, such as cancer cells or normal cells immortalized by introduction of telomerase, can still undergo SIPS in response to oxidative stresses or agents that trigger DNA damage formation such as UV and anticancer drugs like topoisomerases inhibitors.Citation20-22
Figure 1. Senescence and the DNA damage response. Different stimuli can trigger replicative senescence or stress-induced premature senescence through activation of the DNA damage response. Telomere shortening at each DNA replication or alteration of the shelterin complex (e.g., depletion of TRF2, Telomeric repeat-binding factor 2) leads to the unmasking of chromosomes extremities. Oncogene activation leads to deregulated proliferation, promoting replication errors. Oxidative stresses and various other agents can trigger extensive non-telomeric DNA damage.
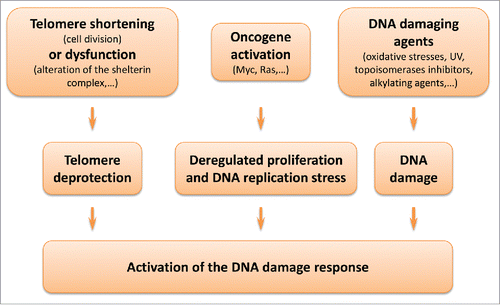
Central to both replicative senescence and SIPS is the activation of the DNA damage response ().Citation10,11,19,20 Development of both stress-induced and telomere-initiated senescence has been associated with activation of the transcription factor and tumor suppressor p53 and its effector p21waf-1. Other major actors of senescence include p16INK4a and pRb. The proteins p21waf-1 and p16INK4A are both cyclin-dependent kinase (CDK) inhibitors. CDK inhibition prevents pRb phosphorylation, and consequently expression of E2F target genes and cell progression into S phase.Citation10,11,19,20
hRSV-induced senescence: Friend or foe?
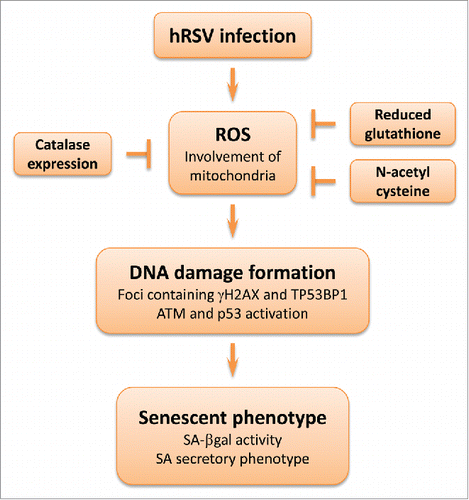
Martinez et al. describe in this issue that hRSV causes in vitro a marked increase of ROS in infected cells, in line with previously published studies. In the two cell lines examined, this oxidative stress produces a strong activation of the DNA damage response as noted by phosphorylation of the ATM (Ataxia Telangiectasia Mutated) kinase, induction of p53 and its effector p21waf-1, as well as presence of γH2AX nuclear foci.Citation6 The antioxidants N-acetyl cysteine and reduced glutathione or overexpression of catalase exert a protective effect. The cascade of molecular events generated ultimately leads to the entry of infected cells into premature senescence (). Interestingly, a similar process seems to occur in vivo: DNA damage (detection of γH2AX) is observed in the airway epithelium of mice infected with hRSV as well as expression of p16.INK4aCitation6
Figure 2. hRSV-induced premature senescence. Cell infection by hRSV leads to generation of ROS, including superoxide anions derived from mitochondria. The oxidative stress is sufficient to trigger DNA damage as noted by the detection of nuclear foci containing TP53BP1 (tumor protein p53 binding protein 1) and γH2AX (in presence of DNA damage, histone variant H2AX is phosphorylated on serine 139 by the ATM kinase or other kinases). Ultimately, cells develop the senescent phenotype. Antioxidants can protect against this process: N-acetyl cysteine, catalase expression or reduced glutathione decrease the oxidative stress as well as DNA damage formation.
As discussed above, the DNA damage response is central to induction of senescence. In this respect, telomere-initiated or stress-induced senescence are tumor suppressor mechanisms that prevent the proliferation of “damaged” cells and constitute a barrier against cancer development. Critically short telomeres are prone to association and favor genomic abnormalities. Stresses that induce SIPS are highly detrimental to cell integrity (oncogene activation, exposure to a DNA damaging agent, oxidative stress) (). Therefore, induction of senescence by hRSV can be seen as a host defense mechanism, instructing “damaged” cells to cease proliferation.Citation10,11,19,20 However, excessive accumulation of senescent cells in a tissue can also have a negative impact on its homeostasis. Senescent cells secrete a number of factors able to modulate cell growth, create a pro-inflammatory micro-environment or remodel the extracellular matrix.Citation9
The airway epithelium contains stem cell populations able to proliferate in response to damage, ensuring repair and thus tissue homeostasis. In the lung ciliated pseudostratified columnar epithelium, basal cells are believed to be pluripotent stem cells able to differentiate in both ciliated cells and non-ciliated secretory cells. In addition, following damage to the ciliated cells, non-ciliated secretory cells can self-renew and differentiate into ciliated cells. In the lung alveolar epithelium, composed of type 1 and type 2 cells, type 2 cells are thought to be the progenitor of type 1 cells.Citation23-27 Moreover, although it was previously thought that ciliated cells were the target of hRSV, recent works indicate that basal stem cells can also be infected.Citation28 Can we accurately demonstrate in vitro and in vivo that hRSV can infect and trigger the senescence of specific airway epithelium stem cell populations? Second, could hRSV infection and subsequent stress-induced senescence lead to a reduced capacity of these populations to self-renew, differentiate and maintain tissue homeostasis? Indeed, somatic stem cells also age and there are examples of decline of their function in different organs. For instance, aged skeletal muscles contain about 50% less satellite cells than healthy adult muscles, indicating a depletion of this somatic stem cell population during organismal aging.Citation29
Further questions
The article of Martinez et al. adds a new layer of complexity to the pathophysiology of hRSV infection.Citation6 Causes and consequences of hRSV-induced senescence could be further investigated. For instance, how hRSV causes an increase of ROS from a mitochondrial origin? Does senescence favors or rather opposes propagation of the infection? And could hRSV-induced senescence be linked to complications sometimes observed in patients?
Furthermore, will the damage alter on the long term the normal functioning of the airway epithelium, or render it more vulnerable to future aggressions? In the article of Martinez et al., it is interesting to note that DNA damage and p16INK4a expression persist in vivo long after the acute phase of the infection, when epithelial cells are negative again for hRSV antigens.Citation6 This suggests that the infection leaves a scar that may have a long-term impact on the function of the tissue. Senescent cells accumulate with age in our tissues, and although this is still intensely debated, there is a growing amount of evidence that it contributes to organismal aging as well as aging-associated pathologies.
A better understanding of the pathophysiology of hRSV infection, including a thorough knowledge of the molecular mechanisms involved, is important for a better management of the disease and hopefully the design of new therapeutic strategies to combat it.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress, COPD, and smokers. Am J Respir Crit Care Med 1996; 154:1055-60; PMID:8887607; http://dx.doi.org/10.1164/ajrccm.154.4.8887607
- Glezen WP, Taber LH, Frank AL, Kasel JA. Risk of primary infection and reinfection with respiratory syncytial virus. Am J Dis Child 1986; 140:543-6; PMID:3706232
- Boeck KD. Respiratory syncytial virus bronchiolitis: clinical aspects and epidemiology. Monaldi Arch. Chest Dis 1996; 51:210-3; PMID:8766196
- Cormier AS, You D, Honnegowda S. The use of a neonatal mouse model to study respiratory syncytial virus infections. Expert Rev Anti Infect Ther 2010; 8:1371-80; PMID:21133663; http://dx.doi.org/10.1586/eri.10.125
- Fleming DM, Elliot AJ, Cross KW. Morbidity profiles of patients consulting during influenza and respiratory syncytial virus active periods. Epidemiol Infect 2007; 135:1099-108; PMID:17291381
- Martinez I, Garcia-Carpizo V, Guijarro T, Garcia-Gomez A, Navarro D, Aranda A, Zambrano A. Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence 2016: 7(4):427-442; PMID:26809688
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25:585-621; PMID:13905658; http://dx.doi.org/10.1016/0014-4827(61)90192-6
- Goldstein S. Replicative senescence: the human fibroblast comes of age. Science 1990; 249:1129-33; PMID:2204114; http://dx.doi.org/10.1126/science.2204114
- Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8:729-40; PMID:17667954; http://dx.doi.org/10.1038/nrm2233
- Saretzki G. Cellular senescence in the development and treatment of cancer. Curr Pharm Des 2010; 16:79-100; PMID:20214620; http://dx.doi.org/10.2174/138161210789941874
- Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192:547-56; PMID:21321098; http://dx.doi.org/10.1083/jcb.201009094
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature 1990; 345:458-60; PMID:2342578; http://dx.doi.org/10.1038/345458a0
- Wright WE, Shay JW. Cellular senescence as a tumor-protection mechanism: the essential role of counting. Curr Opin Genet Dev 2001; 11:98-103; PMID:11163158; http://dx.doi.org/10.1016/S0959-437X(00)00163-5
- Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 2005; 6:611-22; PMID:16136653; http://dx.doi.org/10.1038/nrg1656
- Dumont P, Chen QM, Burton M, Balbeur L, Gonos ES, Frippiat C, Mazarati JB, Eliaers F, Remacle J, Toussaint O. Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic Biol Med 2000; 28:361-73; PMID:10699747; http://dx.doi.org/10.1016/S0891-5849(99)00249-X
- Toussaint O, Dumont P, Dierick JF, Pascal T, Frippiat C, Chainiaux F, Magalhaes JP, Sluse F, Eliaers F, Remacle J. Stress-induced premature senescence (SIPS). Essence of life, evolution, stress and aging. Ann N Y Acad Sci 2000; 908:85-98; PMID:10911950; http://dx.doi.org/10.1111/j.1749-6632.2000.tb06638.x
- Toussaint O, Dumont P, Remacle J, Dierick JF, Pascal T, Frippiat C, Magalhaes JP, Zdanov S, Chainiaux F. Stress-induced premature senescence or stress-induced senescence-like phenotype: one in vivo reality, two possible definitions? How stress, cellular behaviors, growth kinetics and cell heterogeneity interact in senescence. Scientific World J 2002; 2:230-47; http://dx.doi.org/10.1100/tsw.2002.100
- Frippiat C, Dewelle J, Remacle J, Toussaint O. Signal transduction in H2O2-induced senescence-like phenotype in human diploid fibroblasts. Free Radic Biol Med 2002; 33:1334-46; PMID:12419465; http://dx.doi.org/10.1016/S0891-5849(02)01044-4
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell 2007; 130:223-33; PMID:17662938; http://dx.doi.org/10.1016/j.cell.2007.07.003
- Yan Q, Wajapeyee N. Exploiting cellular senescence to treat cancer and circumvent drug resistance. Cancer Biol Ther 2010; 9:166-75; PMID:20118655; http://dx.doi.org/10.4161/cbt.9.3.11166
- te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res 2002; 62:1876-83; PMID:11912168
- de Magalhaes JP, Chainiaux F, Remacle J, Toussaint O. Stress-induced premature senescence in BJ and hTERT-BJ1 human foreskin fibroblasts. FEBS Lett 2002; 523:157-62; PMID:12123824; http://dx.doi.org/10.1016/S0014-5793(02)02973-3
- Evans MJ, Cabral LJ, Stephens RJ, Freeman G. Transformation of alveolar type 2 cells to type 1 cells following exposure to NO2. Exp Mol Pathol 1975; 22:142-50; PMID:163758; http://dx.doi.org/10.1016/0014-4800(75)90059-3
- Evans MJ, Cabral-Anderson LJ, Freeman G. Role of the Clara cell in renewal of the bronchiolar epithelium. Lab Invest 1978; 38:648-53; PMID:661220
- Evans MJ, Shami SG, Cabral-Anderson LJ, Dekker NP. Role of nonciliated cells in renewal of the bronchial epithelium of rats exposed to NO2. Am J Pathol 1986; 123:126-33; PMID:3963147
- Hong KU, Reynolds SD, Watkins S, Fuchs E, Stripp BR. In vivo differentiation potential of tracheal basal cells: evidence for multipotent and unipotent subpopulations. Am J Physiol Lung Cell Mol Physiol 2004; 286:L643-649; PMID:12871857; http://dx.doi.org/10.1152/ajplung.00155.2003
- Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, Wang F, Hogan BLM. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 2009; 4:525-34; PMID:19497281; http://dx.doi.org/10.1016/j.stem.2009.04.002
- Persson BD, Jaffe AB, Fearns R, Danahay H. Respiratory syncytial virus can infect basal cells and alter human airway epithelial differentiation. PloS One 2014; 9:e102368; PMID:25033192; http://dx.doi.org/10.1371/journal.pone.0102368
- Jung Y, Brack AS. Cellular mechanisms of somatic stem cell aging. Curr Top Dev Biol 2014; 107:405-38; PMID:24439814; http://dx.doi.org/10.1016/B978-0-12-416022-4.00014-7