
ABSTRACT
Blue and green molds, the common phenotypes of post-harvest diseases in fruits, are mainly caused by Penicillium fungal species, including P. italicum, P. digitatum, and P. expansum. We sequenced and assembled the genome of a P. italicum strain, which contains 31,034,623 bp with 361 scaffolds and 627 contigs. The mechanisms underlying the evolution of host specificity among the analyzed Penicillium species were associated with the expansion of protein families, genome restructuring, horizontal gene transfer, and positive selection pressure. A dual-transcriptome analysis following the infection of Valencia orange (Citrus sinensis) by P. italicum resulted in the annotation of 9,307 P. italicum genes and 24,591 Valencia orange genes. The pathogenicity of P. italicum may be due to the activation of effectors, including 51 small secreted cysteine-rich proteins, 110 carbohydrate-active enzymes, and 12 G protein-coupled receptors. Additionally, 211 metabolites related to the interactions between P. italicum and Valencia orange were identified by gas chromatography-time of flight mass spectrography, three of which were further confirmed by ultra-high performance liquid chromatography triple quadrupole mass spectrometry. A metabolomics analysis indicated that P. italicum pathogenicity is associated with the sphingolipid and salicylic acid signaling pathways. Moreover, a correlation analysis between the metabolite contents and gene expression levels suggested that P. italicum induces carbohydrate metabolism in Valencia orange fruits as part of its infection strategy. This study provides useful information regarding the genomic determinants that drive the evolution of host specificity in Penicillium species and clarifies the host-plant specificity during the infection of Valencia orange by P. italicum.
IMPORTANCE
P. italicum GL_Gan1, a local strain in Guangzhou, China, was sequenced. Comparison of the genome of P. italicum GL_Gan1 with other pathogenic Penicillium species, P. digitatum and P. expansum, revealed that the expansion of protein families, genome restructuring, HGT, and positive selection pressure were related to the host range expansion of the analyzed Penicillium species. Moreover, gene gains or losses might be associated with the speciation of these Penicillium species. In addition, the molecular basis of host-plant specificity during the infection of Valencia orange (Citrus sinensis) by P. italicum was also elucidated by transcriptomic and metabolomics analysis. The data presented herein may be useful for further elucidating the molecular basis of the evolution of host specificity of Penicillium species and for illustrating the host-plant specificity during the infection of Valencia orange by P. italicum.
Introduction
Host specificity refers to the ability of an organism to colonize another organism [Citation1]. Some plant pathogens have a broad host range, whereas others can only colonize specific plant species or families. Even within the same genus, there are considerable differences in the host range among species, which may be related to pathogen evolution. During the evolution of plant pathogens, host shifts may occur to broaden or limit the host range [Citation2]. In extreme cases, a pathogen evolves to colonize a new host that is phylogenetically distant from the original hosts (i.e., host jump), possibly resulting in the emergence of new fungal diseases [Citation3,Citation4]. Therefore, elucidating the molecular basis of the evolution of host specificity in fungal plant pathogens is critical for developing strategies to decrease the yield losses of economically valuable crops.
The genus Penicillium comprises ascomycetous fungal species that are widely distributed, with important implications for natural environments as well as in the food and drug industries. Some members of this genus produce penicillin, an antibiotic, whereas other species are used to make cheese. However, some Penicillium species are among the most important plant pathogens responsible for postharvest diseases. For example, Penicillium italicum, Penicillium digitatum, and Penicillium expansum cause losses of up to 10% of harvested crops. Of these species, P. italicum and P. digitatum exclusively infect citrus fruits, but P. expansum can infect diverse fruit and vegetable crops, such as apple, pear, peach, strawberry, and tomato, but not citrus fruits [Citation5]. The genomes of P. italicum, P. digitatum, and P. expansum strains have been sequenced [Citation6–Citation9]. Additionally, Li et al. [Citation8] and Ballester et al. [Citation7] focused on the relationship between secondary metabolism and infections in P. expansum. However, the pathogenicity and evolution of host specificity in Penicillium species have remained largely uncharacterized.
The evolution of host specificity of fungi is mediated by a complex process involving multiple genes and mechanisms. A multidisciplinary approach may unveil new molecular determinants that drive the evolution of host specificity. In this study, we sequenced the genome of a P. italicum strain and then applied comparative genomics, dual-transcriptomics, and metabolomics analyzes to uncover the genomic determinants associated with the evolution of Penicillium species and elucidate the molecular mechanism underlying the host-plant specificity during the infection of Citrus sinensis by P. italicum. The results presented herein provide the molecular basis for clarifying the genomic determinants that drive the evolution of host specificity of Penicillium species and the host-plant specificity during the infection of Valencia orange by P. italicum.
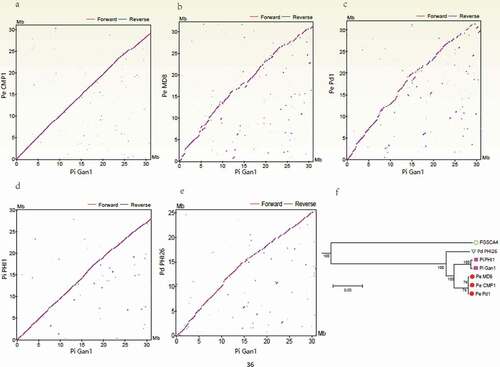
Figure 1. Comparison of the Penicillium italicum, P. digitatum, and P. expansum genomes. (a – e) Dot-plot analysis of the pairwise alignments of the P. italicum GL_Gan1 genome and the genomes of the other analyzed Penicillium species. (f) A phylogenetic tree revealing the relationships among the following Penicillium species and strains: P. italicum (Gan1 and PHI1), P. expansum (CMP1, MD8, and Pd1), and P. digitatum (PHI26).
Results and Discussion
Penicillium italicum genome sequencing and annotation
An Illumina HiSeq 2000 sequencing platform was used to generate 1,815 Mb of raw data for the P. italicum strain GL_Gan1 genome. The assembled genome contained 31,034,623 bp, with an average GC content of 45.83%, including 361 scaffolds ranging from 1,002 bp to 994,651 bp and 627 contigs ranging from 213 bp to 867,867 bp. The N50 values of the scaffolds and contigs were 316.59 kb and 196.54 kb, respectively (), which were considerably higher than those of the released genomes of P. italicum B3 and P. italicum PITC [Citation7,Citation8]. Moreover, the P. italicum GL_Gan1 genome comprised 9,447 genes, which consisted of 15,524,911 bp, including 13,960,971 bp of exons and 1,563,940 bp of introns. The repeat sequence contained 3,791,084 bp. The predicted genes included 7,136 core genes and 85 species-specific genes (Dataset S1), which represented 75.54% and 0.90% of the total number of genes, respectively. The genes were functionally annotated with the Gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Clusters of Orthologous Groups (COG) databases (Fig. S1). The top matches for the GO, KEGG, and COG databases included 3,350 (35.46%), 1,172 (12.41%), and 1,447 (15.31%) genes, respectively, and the most abundant assigned terms were ‘metabolic process’, ‘xenobiotic bio-degradation and metabolism’, and “general function prediction only”, respectively (Fig. S1).
Table 1. Penicillium italicum GL_Gan1 genome properties compared with those of other sequenced P. italicum strains.
Horizontal gene transfer (HGT) is a major evolutionary factor that enables organisms to quickly acquire new genes that enhance environmental adaptation and survival. In this study, we identified 383 transferred genes in the P. italicum GL_Gan1 genome (Dataset S1). Regarding the functional annotation with the COG database (Dataset S1), the top three categories were secondary metabolite biosynthesis, transport, and catabolism (49 proteins), amino acid transport and metabolism (48 proteins), and energy production and conversion (44 proteins). Similarly, the top categories for the functional annotation with the GO database (Dataset S1) were metabolic process (294 genes) and catalytic activity (291 genes). Interestingly, the most enriched KEGG pathways (Dataset S1) were xenobiotics biodegradation and metabolism (137 genes), amino acid metabolism (104 genes), and carbohydrate metabolism (97 genes). These results suggested that genes associated with metabolic processes and/or products are prone to be horizontally transferred [Citation10]. Moreover, GL_Gan1_GLEAN_10000555 encodes a polyketide synthase (PKS) with two active domains (MDR and Qor). This PKS is a core enzyme responsible for catalyzing the first reaction during the biosynthesis of secondary metabolites (SMs) [Citation11]. We constructed a phylogenetic tree based on PKS sequences from Aspergillaceae, Bacteria, Glomerellaceae, Nectriaceae, Clavicipitaceae, and so on. There were three major clades, Polyporaceae and bacteria, Aspergillaceae and others. Surprisingly, when PKS from seven Bacteria genes and seven Penicillium genes were constructed a phylogenetic tress, it was found that Paenibacillus mucilaginosus gene was clustered with Penicillium genes (Fig. S2). Paenibacillus mucilaginosus is able to promote crop growth and widely used as microbial fertilizer in China [Citation12]. Possibly, horizontal gene transfer occurred for P. italicum PKS gene from Paenibacillus mucilaginosus, however, further experiment is required to verify the hyposthesis. A similar HGT was reported for the phytohormone biosynthetic genes in Fusarium species [Citation13].
Plants interact with pathogens through microbial effectors and host immune receptors. The successful colonization of a plant pathogen is the result of the interaction between pathogen effectors and host immune receptors. Pathogen effectors repress the host immune response to facilitate pathogen growth and colonization, which are critical for the pathogenicity of filamentous fungi. Sanchez-Vallet et al. proposed that some small-size peptides for secretion, cell wall-degrading enzymes, protease inhibitors, interactors with the ubiquitin-proteasome system, and disruptors of the hormone signaling pathway can act as effectors [Citation14]. It has been reported that some fungal effector proteins identified are relatively small protein containing fewer than 200 amino acids and more than 2% cysteines, i.e. small secreted cysteine-rich proteins (SSCPs), constituting a common source of fungal effectors [Citation15]. In the present study, a total of 444 putative effector genes were identified in the P. italicum GL_Gan1 genome, of which 51 genes encoding SSCPs (Dataset S1) possibly play roles in trigger resistance or susceptibility in citrus fruit [Citation16].
Plant cell wall is a complex matrix of polysaccharides, including pectin, hemicellulose and cellulose, which provides mechanical support for cells and forms a barrier against fungal invasion. Plant pathogens are equipped with multiple carbohydrate-active enzymes (CAZymes), including glycoside hydrolases (GHs), glycosyl transferases (GTs), polysaccharide lyases (PLs), carbohydrate esterases (CEs) and carbohydrate-binding modules (CBMs), which facilitate fungal invasion via degradation or modification of plant cell walls [Citation17,Citation18]. P. italicum is a necrotrophic pathogen and only infects citrus fruit through peel injuries in the field, the packing house, or during commercialization chains [Citation6]. Initiative infection of P. italicum is usually seen only after about 3 d of incubation at room temperature, with water-soaked and soft peel. Therefore, hydrolytic enzymes produced by P. italicum appear responsible for the maceration of the tissue during disease development [Citation19]. In the present study, 1,290 genes encoding CAZymes were identified in the P. italicum genome, including 18 PLs, 383 GTs, 623 GHs, 141 CEs, and 274 CBMs, which was much more than that observed in the other published and annotated P. italicum genome [Citation8]. The number of CAZymes in P. italicum was similar to that of the necrotrophic fungi Fusarium oxysporum (approximately 1200), but much more than those of saprotrophic fungi, such as Chaetomium thermophilum var. thermophilum DSM 1495 and Neurospora crassa OR74A (less than 600) [Citation17]. We also noticed that GHs were the most abundant CAZymes in P. italicum genome, of which some subgroups, including GH76, GH78, GH92, GH3, GH16, GH51, GH55, GH43, GH5, GH2, GH17, GH71, were involved in degradation of cell wall polysaccharides (www.cazy.org). The distribution of CAZymes in P. italicum was similar with other necrotrophic fungi, such as Fusarium oxysporum, Fusarium verticillioides, Nectria haematococca, Alternaria brassicicola, Parastagonospora nodorum, and Corynespora cassiicola [Citation17].
Comparative genomics analysis reveals the evolution of Penicillium species
To elucidate the evolution of host specificity and the pathogenicity of Penicillium species, the genomes of two P. italicum strains (GL_Gan1 and PHI1/PITC), one P. digitatum strain (PHI26), and three P. expansum strains (CMP1, Pd1, and MD8) were compared. A genome wide analysis of synteny showed that Pe_CMP1/Pi_Gan1 () and Pi_PHI1/Pi_Gan1 () had high levels of synteny, followed by Pd_PHI26/Pi_Gan1 (). A total of 7136 single-copy core genes were used to constructed phylogenetic tree (), with Aspergillus nidulans_FGSCA4 as an outgroup species, to further elucidate the evolutionary relationships among P. italicum, P. expansum, and P. digitatum. The results showed that P. italicum Gan1 and PHI1 were cluster into one group, while P. expansum CMP1, Pd1, and MD8 were cluster into another group. Moreover, the phylogenetic distance between P. italicum Gan1 and PHI1 was shorter than those between PHI26 and P. expansum CMP1, Pd1, and MD8. The inconsistence between synteny and phylogenetic tree might be related to the application of genome and single-copy core genes for comparative analysis, respectively. However, the comparative analysis of core genome showed the close relationship between P. italicum and P. expansum. Our results were consistent with the species tree reconstructed using 524 single-copy genes present in seven Penicillium species [Citation7] and 2,134 single-copy genes present in eight Penicillium species [Citation20].
The comparative genomics analysis demonstrated the evolutionary plasticity of these Penicillium genomes. The core genomes comprised 7,690 genes (P. digitatum), 8,375 genes (mean) (P. italicum), and 9,342 genes (mean) (P. expansum) (), suggesting that lineage-specific gene expansion was essential for P. expansum evolution and its adaptations to diverse hosts. Similar results were reported by Yoshida et al. [Citation21] who proposed that gene gains and losses are two of the primary contributors to the genome shift essential for species evolution.
Table 2. Core and strain-specific genes in the Penicillium italicum, P. digitatum, and P. expansum genomes.
The expansion of protein families associated with fungal pathogenicity was speculated to force the speciation of these Penicillium species. presents the differences in the protein content among Penicillium species. 6,309 and 6851 protein families were detected in P. digitatum and P. italicum, respectively, whereas 7,640 protein families were detected in P. expansum, implying that the expansion of protein families has occurred from P. digitatum and P. italicum to P. expansum, which might be related to the fungal pathogenicity of the three Penicillium species.
Table 3. Comparison of protein family number associated with fungal pathogenicity among Penicillium species.
Pathogen-host interaction (PHI) are crucial for initiating the infections of a host by a pathogen, which is regulated by pathogenesis-related secreted proteins, such as CAZymes and SSCPs [Citation22]. PHI-base database (http://www.phibase.org/) is to provide expertly curated molecular and biological information on genes proven to affect the outcome of pathogen-host interactions. In the present study, searching against the PHI database identified 1728 putative PHI genes in the P. italicum GL_Gan1 genome which were homologous genes involved in pathogenicity in other fungi. A comparison of the number of PHI genes revealed that P. digitatum (average 1,503) has fewer PHI genes than P. expansum (average 2,214) and P. italicum (average 1,728) (). Furthermore, P. italicum, P. digitatum and P. expansum shared common subgroups (399) with differential numbers of PHI genes in some subgroups. Moreover, each species had its own specific PHI subgroups (Dataset S2). Possibly, the differential compositions and number in PHI genes among three Penicillium species were related to the different host ranges.
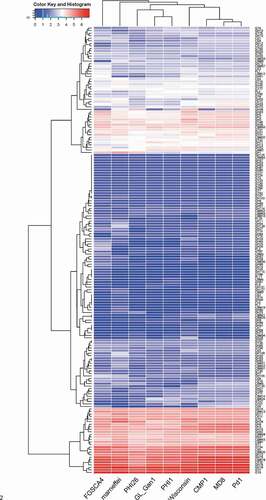
CAZymens play important role in the invasion of pathogens on host plants by degrading cell wall polysaccharides, such as cellulose, hemicellulose and pectin [Citation17]. In pathogens, GTs are mainly involved in carbohydrate assembly, while GHs, CEs and PLs are cell wall degradation-related enzymes [Citation23]. Among P. italicum, P. expansum, and P. digitatum, P. expansum has the most CAZymes, followed by P. italicum (). We constructed a heat map for the different CAZyme subgroups to further elucidate the evolutionary relationships among P. italicum, P. expansum, and P. digitatum. There were no obvious differences in numbers in most of the CAZyme subgroups among the three analyzed species. However, significant differences were detected for the subgroups involving GH76, GH78, GH79, GH92, GH3, GH16, GH51, GH55, GH32, GH75, GH43, GH5, GH2, CE10, CE9, CE15, GT34, CBM18, CBM13, CBM12, CBM50 (), with P. expansum harboring more of these families than P. italicum and P. digitatum. GH92 s and GH76 s are exo-acting α-mannosidases and endo-acting α-mannanases, respectively [Citation24,Citation25], while GH3s and GH43 s are xylan degradation-related enzymes (www.cazy.org), which could together play important roles in degrading hemicellulose and softening host tissues. The majority of GH16, GH51, GH55 and GH5 CAZymes are endoglucanase, lichenase/endo-β-1,3–1,4-glucanase, endo-β-1,4-glucanase, xyloglucanase, endo-β-1,4-xylanase, glucan 1,3-β-glucosidase, glucan endo-1,3-β-D-glucosidase, endo-β-1,4-xylanase, β-glucosidase (www.cazy.org), which are important enzymes involving in the degradation of cellulose and hemicellulose. GH78 s are rhamnogalacturonan α-L-rhamnohydrolases that could also participate in host cell wall degradation [Citation23]. Lipase is an emerging research field on virulence mechanisms in fungi. CE10 s belong to lipases which hydrolyze carboxyl ester bonds on triacylglycerols, and may act against host morpho-anatomical barriers (wax and cuticle) and be involved in fungal pathogenesis [Citation17,Citation23]. CE15 s are 4-O-methyl-glucuronoyl methylesterase (Glucuronoyl esterase), which is considered to disconnect hemicellulose from lignin through the hydrolysis of the ester bond between 4-O-methyl-D-glucuronic acid residues of glucuronoxylans and aromatic alcohols of lignin [Citation26]. The greater amount of cell wall polysaccharide degradation-related enzymes in P. expansum might be beneficial for degradation of cell walls and also implied greater pathogenicity.
Figure 2. Heat map of CAZyme genes reveals the evolutionary relationships among Penicillium italicum, P. digitatum, and P. expansum. Several other fungi, Aspergillus nidulans_FGSCA4, Penicillium rubens Wisconsin 54–1255 and Penicillium marneffei, also were included in the heat map.
Small secreted cysteine-rich proteins (SSCPs) are important fungal effectors that trigger resistance or susceptibility in specific host plants [Citation15]. These proteins can recognize the products of plant R genes and are related to disease symptom development and fungal pathogenicity. The P. expansum genome carries an average of 70 SSCP genes, whereas the P. italicum (average 57) and P. digitatum (average 61) genomes consist of fewer SSCP genes.
G protein-coupled receptors (GPCRs) are seven-transmembrane domain-containing receptors that transduce extracellular signals and activate intracellular responses [Citation27]. They have important functions related to fungal growth and survival. The number of GPCRs varies depending on the fungal species. For example, the Sporisorium scitamineum genome encodes only six GPCRs, whereas the Magnaporthe grisea genome includes 61 GPCR genes [Citation28,Citation29]. Penicillium species have a few GPCRs, and no GPCR gene expansion was detected during evolution.
Cytochrome P450 s (P450 s) belong to a superfamily of mono-oxygenases, which are distributed in a wide range of organisms. The diversity in the number of P450 genes and families among organisms reflects an evolutionary driving force toward speciation [Citation30]. Compared with the number of P450 genes in P. italicum (average 29) and P. digitatum (average 12), a marked gene expansion occurred in P. expansum (average 43).
Secondary metabolites are necessary for fungi to produce a variety of chemical compounds, including toxins and antibiotics, which contribute to fungal virulence, host specificity, and adaptations to ecological niches [Citation31]. The specialist P. digitatum has fewer SM biosynthetic gene clusters (average 32) than P. italicum (average 43) and P. expansum (average 65). Recently Wu et al. (2019) reported the similar composition of SM gene clusters in different Penicillium species, but further functional characterization of all these genes are required to be exactly identified by RNA interference and/or gene deletion studies [Citation32].
Therefore, protein-family expansions in these Penicillium lineages were possibly associated with the evolutionary processes in these species. It seems that P. italicum is a transitional species in the Penicillium lineage that developed during the evolution of P. digitatum to P. expansum. Thus, P. expansum may be a newer species than P. italicum. This is consistent with the principle of speciation that suggests the newly evolved P. expansum should have a broader host range than the older related species. Additionally, our data are consistent with those of previous studies on seven Metarhizium species and three Fusarium species, which indicated that the genome and protein families expanded considerably in the transitional and generalist species [Citation33,Citation34]. Alternatively, these Penicillium species may have undergone a parallel evolution, but the divergence of P. italicum and P. digitatum occurred because of gene losses, particularly the loss of genes associated with virulence and metabolic products, resulting in major changes to P. italicum and P. digitatum that enabled them to infect new hosts. Accordingly, these two species may have been exposed to extreme external pressures that led to a rapid evolution of effectors for their colonization of a new host species. Similarly, a loss of genes associated with virulence and toxin biosynthesis in host-specific strains of Metarhizium species resulted in the divergence or rapid evolution of specialist Metarhizium species [Citation35]. Moreover, gene losses rather than gains occurred in Melanopsichium pennsylvanicum, which expanded its host range from monocot to dicot hosts [Citation36].
Genome restructuring may have contributed to the evolution of host specificity. Transposable elements (TE) cause gene mutations and the evolution of genomes [Citation37]. We detected considerably more TEs in P. digitatum and P. italicum than in P. expansum (Table S1), implying that P. expansum has fewer mutations than P. digitatum and P. italicum after the reinforcement of reproductive isolation. Moreover, the plasticity of the P. digitatum and P. italicum genomes was greater than that of the P. expansum genome.
Using Ka/Ks > 1 and P < 0.01 as criteria, we detected 17 genes under positive selection pressure among the examined Penicillium species. Positively selected genes, most of which are related to fungal virulence (Dataset S1), were frequently detected between P. italicum and P. expansum, rather than in P. italicum itself or between P. italicum and P. digitatum. These results imply that these genes may have contributed to the divergence of the host specificity between P. italicum and P. expansum, thereby providing important evidence of the evolutionary processes of these Penicillium species as well as the adaptation of P. expansum to various hosts.
Dual-transcriptome analysis of P. italicum GL_Gan1 and Valencia orange during colonization
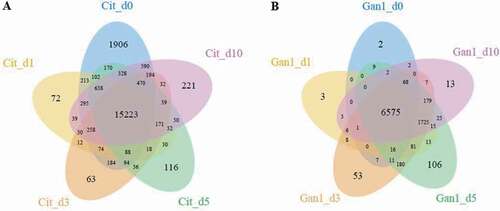
To explore the mechanism underlying the colonization and pathogenicity of P. italicum on Citrus sinensis, the transcriptome profiles of Valencia orange and P. italicum GL_Gan1 during the colonization were analyzed. We constructed RNA-sequencing (RNA-seq) libraries for the inoculated Valencia orange at 0, 1, 3, 5, and 10 days post-inoculation (dpi). The libraries were sequenced with the Illumina RNA-seq technology, and more than 20 M reads were generated for each library (Table S2). Additionally, 0.61%–57.27% of the clean reads were mapped to the P. italicum genome, in which 6,840–9,145 expressed genes were detected. Moreover, 12.07%–82.82% of the clean reads were mapped to the C. sinensis genome, in which 17,610–21,896 expressed genes were detected (Table S2). Furthermore, 6575 and 15,223 genes were expressed at all stages in P. italicum and Valencia orange, respectively (). Compared with the expression levels at 0 dpi, 228, 414, 376, and 336 genes in P. italicum and 301, 1,314, 1,955, and 479 genes in Valencia orange were differentially expressed at 1, 3, 5, and 10 dpi, respectively.
Figure 3. Venn diagram presenting the number of genes expressed in Valencia orange (a) and P. italicum (b) on days 0, 1, 3, 5, and 10 after the inoculation.
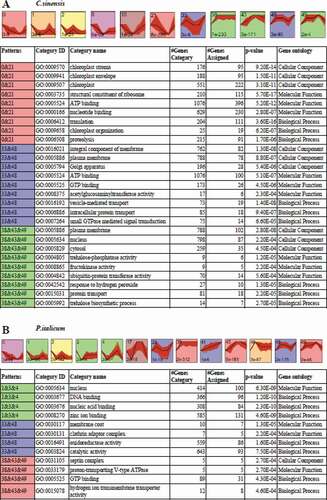
A short time-series expression miner (STEM) clustering analysis of gene expression during the infection of Valencia orange by P. italicum revealed that the fruit responses to P. italicum included cell wall and membrane degradation, accelerated polysaccharide metabolism and energy transformation, and increased metabolite production (). The genes associated with cellular components that exhibited the same temporal expression patterns were clustered together. Among these genes, the expression levels of the genes associated with the chloroplast, including stroma (95 assigned genes), envelope (95 assigned genes), thylakoid (17 assigned genes), and organization (19 assigned genes), were significantly down-regulated, suggesting that the chloroplast of the infected Valencia orange may have degraded or was otherwise adversely affected. Similarly, the expression levels of many genes related to protein synthesis, including the structural constituents of ribosomes (115 assigned genes) and translation (111 assigned genes), were down-regulated in the inoculated Valencia orange, indicating the infection impeded protein synthesis (). Additionally, glucosyltransferases and the related metabolism may be activated in Valencia orange. The expression levels of five of nine genes associated with fructokinase activity were up-regulated. It is possible that the growth of P. italicum exhibits a preference for glucose rather than fructose as an energy source. The impact of fungal pathogen on plant host transcriptome have widely revealed the altered gene expression patterns involved in defense and stress responses, cell wall structure, chloroplast biogenesis, carbon metabolism and transportation, and biosynthesis of secondary metabolites [Citation38,Citation39].
Figure 4. STEM clusters of gene expression profiles of P. italicum and C. sinensis during the colonization of P. italicum in C. sinensis. (a) Top panel: eleven representative temporal gene expression patterns were identified in C. sinensis; Colored backgrounds were used to differentiate different possible model profiles recognized by STEM, with the number of clustered genes indicated in the top left corner, the bottom left presents the p-value of number of genes assigned versus expected. Bottom panel: GO enrichment of three clusters of expression patterns genes category with top three of ontology, genes category, genes assigned and the significance (p-value<0.001) were listed. (b) Top panel: thirteen representative temporal gene expression patterns were identified in P. italicum; Colored backgrounds were used to differentiate different possible model profiles recognized by STEM, with the number of clustered genes indicated in the top left corner, the bottom left presents the p-value of number of genes assigned versus expected. Bottom panel: GO enrichment of three clusters of expression patterns genes category with top three of ontology, genes category, genes assigned and the significance (p-value<0.001) were listed.
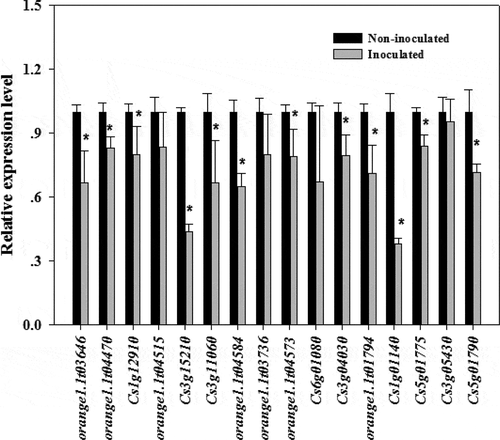
Plants have evolved sophisticated strategies to escape various pathogen attacks. Plant resistance genes (R genes) are important for resisting pathogen infections. These genes are structurally conserved in vertebrates and plants, with the following typical domains: NBS, LRR, TIR, and CC. A total of 636 R genes were identified in the Valencia orange genome (Table S3). The expression of most of these genes was down-regulated during infection (). We randomly selected 16 down-regulated R genes from the transcriptome data and compared their expression in non-inoculated and inoculated Valencia orange. Of the 16 genes, the expression of 12 was significantly lower in the inoculated Valencia orange than in the non-inoculated fruit at 3 dpi (). These results indicated that the P. italicum infection adversely affected R gene-based immunity. Houterman et al. [Citation40] reported that Fusarium oxysporum secretes an effector, Avr1, which can induce and suppress R gene-based immunity in infected tomato plants. This effector activates the disease resistance of tomato plants via R genes (I or I-1), but suppresses the protective effects of two other R genes, I-2 and I-3.
Figure 5. Heat map for the expression profiles of Valencia orange R genes during the infection by P. italicum. Blue and red bars represent low and high expression levels based on the number of transcripts, respectively.
Figure 6. Comparison of the expression levels of R genes in P. italicum-infected and healthy peel of Valencia orange at 3 dpi. * indicates significant difference (p < 0.05).
Our STEM clustering analysis also confirmed that P. italicum exhibited accelerated growth and development, as the expression levels of all five genes associated with a cell division control-related protein were up-regulated during infection. Similarly, many genes related to ATP hydrolysis also exhibited up-regulated expression (). Therefore, P. italicum may be very actively absorbing nutrients, leading to accelerated growth.
P. italicum secretes many proteins and substances that enable it to infect and colonize Citrus sinensis, such as CAZymes, SSCPs, GPCR, and SMs. The CAZymes degrade plant cell walls to create an entry point in the plant host. In the present study, 110 genes encoding CAZymes were differentially expressed in P. italicum during infection, including genes belonging to the GH28, GH78, GH95, GH105, CE8, CE12, PL1, PL3, and PL4 families. These CAZymes help degrade the pectin, hemicellulose, and cellulose in plant cell walls. Specifically, the expression levels of several genes encoding CAZymes, including GL_Ganl_CLEAN_10003242, GL_Ganl_CLEAN_10005653, GL_Ganl_CLEAN_10005681, GL_Ganl_CLEAN _10005669, GL_Ganl_CLEAN_10000302, and GL_Ganl_CLEAN_10007363, were substantially up-regulated in P. italicum during the infection of Valencia orange (). Thus, these genes may be critical for the infection. Ballester et al. [Citation7] reported that a large number of gene encoding cell wall-degrading enzymes including polygalacturonase, pectate lyase, glycosyl hydrolase and xylanase, are obviously up-regulated in P. expansum when infecting apple fruit at 2–3 dpi. The SSCPs represent a common source of fungal effectors [Citation41]. In the current study, 51 genes encoding SSCPs were differentially expressed in P. italicum during infection (Dataset S3). The expression of a number of SSCP genes was down-regulated in P. italicum during infection. Additionally, 15 genes encoding SSCPs were not expressed at 0 dpi, but were expressed at 1 or 3 dpi. Moreover, the expression levels of four SSCP genes (GL_Gan1_GLEAN_10002725, GL_Gan1_GLEAN_10004237, GL_Gan1_GLEAN _10005638, and GL_Gan1_GLEAN_10001595) increased considerably during the early infection stage, implying these SSCPs influence P. italicum pathogenicity. Similarly, Bradshaw et al. [Citation42] examined the gene expression dynamics of Dothistroma septosporum when it invades its host, and revealed the up-regulated expression of genes mainly encoding SSCPs, CAZymes, and SMs in three infection cycle stages. Alkan et al. [Citation43] simultaneously analyzed the transcriptomes of Colletotrichum gloeosporioides and tomato fruits during their interaction, and determined that many SSCP genes are expressed in different stages. Overall, the RNA-seq results highlighted the arsenal of putative pathogenicity factors and identified specific candidates that should be functionally analyzed to further characterize their effect on P. italicum pathogenicity.
Figure 7. CAZyme genes and their expression levels during the infection of Valencia orange by P. italicum GL_Gan1.
Metabolomics analysis of non-infected and infected Valencia orange by P. italicum
We used GC-TOF-MS to analyze the metabolome of the cuticle tissues in non-infected and infected Valencia orange fruits at 3 dpi. A total of 592 metabolites were identified and quantified, of which 207 metabolites had the similarity over 700, including sugars, polyols, organic acids, amino acids, and SMs (Table S4). The abundance of 51 metabolites was significantly different between the inoculated and non-inoculated Valencia orange fruits (). Most of the differentially accumulated metabolites are related to carbohydrate metabolism, amino acid metabolism, fatty acid metabolism, secondary substance metabolism, and nucleic acid metabolism.
Table 4. Identification of differentially accumulated metabolites (p < 0.05) between non-infected and infected Valencia orange fruits.
Trehalose is a widely distributed non-reducing disaccharide that has important effects on plant growth and development as well as responses to abiotic stresses [Citation44]. There is emerging evidence that trehalose is also involved in plant responses to pathogens. For example, exogenous trehalose acts as an elicitor to induce the resistance of wheat to powdery mildew disease [Citation45]. Zhang et al. [Citation46] applied virus-induced gene silencing to reveal the involvement of several putative trehalose-6-phosphate synthase/phosphatase genes in the resistance of tomato plants to Botrytis cinerea and Pseudomonas syringae. In the present study, the trehalose concentration of non-infected Valencia orange was 3.45 times higher than that in infected fruits at 3 dpi (). The decreased accumulation of trehalose in infected Valencia orange was also confirmed by ultra-high performance liquid chromatography and triple quadrupole mass spectrometry (UPLC-QQQ-MS) (). The Valencia orange genome carries six genes (Cs3g05430, orange1.1t05805, Cs5g01775, Cs5g01780, Cs7g22230, and Cs5g01790) encoding trehalase, which is the enzyme responsible for the degradation of trehalose [Citation47]. Of these genes, Cs3g05430, orange1.1t05805, Cs5g01775 and Cs7g22230 were more highly expressed in the infected Valencia orange than in the non-infected fruit at 3 dpi, whereas there were no significant differences in the expression of Cs5g01780 and Cs5g01790 (). In addition, we identified six trehalose-phosphate synthase genes (GL_Gan1_GLEAN_10003645, GL_Gan1_GLEAN_ 10,003,647, GL_Gan1_GLEAN_10007719, GL_Gan1_GLEAN_10006325, GL_Gan1_GLEAN _10009252, GL_Gan1_GLEAN_10007138) and one trehalase gene (GL_Gan1_GLEAN_10009387) in P. italicum GL_Gan1. Among these genes, expression of GL_Gan1_GLEAN_10003645 were significantly down-regulated at 3 dpi compared with at 0 dpi, while expression of other genes showed no significant difference between at 0 dpi and at 3 dpi. The up-regulated expression of trehalase genes in Valencia orange and the down-regulated expression of trehalose-phosphate synthase in P. italicum was consistent with the decreased accumulation of trehalose in the infected Valencia orange fruit. Therefore, during the infection of Valencia orange, P. italicum induces the expression of trehalose degradation-related genes, which accelerates trehalose degradation and decreases disease resistance.
Figure 8. (a) Comparison of the concentrations of specific chemicals (trehalose, salicylic acid, and D-sphingosine) between the P. italicum-infected and healthy cuticles of Valencia orange based on a UPLC-QQQ-MS analysis. Expression of six genes encoding trehalase (b) and three genes encoding serine palmitoyl transferase (c) in non-inoculated and inoculated Valencia orange fruits at 3 dpi.
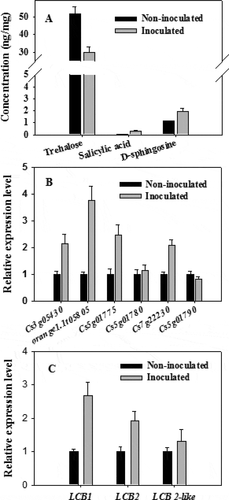
Salicylic acid is a key plant hormone that mediates host responses to microbial pathogens. A UPLC-QQQ-MS analysis indicated the salicylic acid concentration was 0.27 ng/mg in inoculated fruits and 0.055 ng/mg in non-inoculated fruits at 3 dpi, implying that a P. italicum infection induces salicylic acid biosynthesis in Valencia orange. Sphingosine is an important regulator of SA accumulation in plant cells [Citation48]. Additionally, D-sphingosine levels in non-inoculated Valencia orange was significantly lower than that in inoculated Valencia orange (), which was consistent with the UPLC-QQQ-MS data (). Serine palmitoyl transferase, a heterodimer composed of LCB1 and LCB2 subunits, catalyzes the first reaction of the biosynthesis of LCB (sphingoid long-chain base) [Citation48]. The LCB1, LCB2, and LCB2-like genes were expressed more highly in inoculated Valencia orange than in non-inoculated Valencia orange (). This explains the high D-sphingosine content in the inoculated Valencia orange. These results suggest that the infection of Valencia orange by P. italicum involves the signaling pathways associated with sphingolipid metabolism and SA accumulation.
The abundance of several disease resistance-related metabolites, including galactinol, ferulic acid, and benzoic acid, significantly decreased in the inoculated Valencia orange at 3 dpi, which likely contributed to the susceptibility of the fruits to a P. italicum infection [Citation49,Citation50].
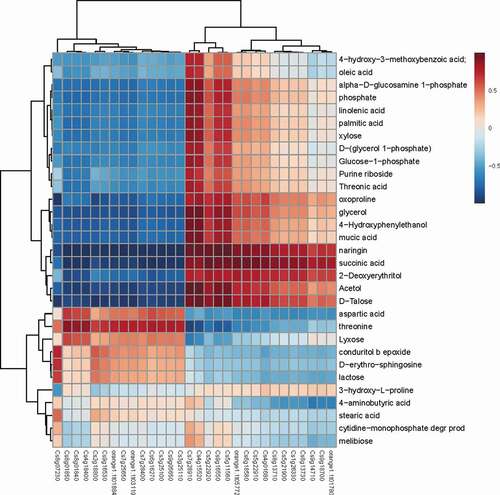
An analysis of the correlation between metabolite accumulation and gene expression clarified the role of primary or secondary metabolism in the disease resistance of Valencia orange or the pathogenicity of P. italicum. The expression levels of several carbohydrate metabolism-related genes, including Cs7g28910 (UDP-apiose/xylose synthase), Cs4g15520 (hexokinase), Cs5g11560 (UDP-glucose 6-dehydrogenase), Cs5g22920 (fructokinase), and Cs9g16550 (fructokinase), were positively correlated with the accumulation of metabolites (), indicating that carbohydrate metabolism was related to the infection by P. italicum.
Figure 9. Heat map analysis of metabolite contents and gene expression profiles. Correlation of metabolite content (vertical axis) and gene expression (horizontal axis) between P. italicum-infected and healthy cuticles of Valencia orange at 3 dpi. Red and blue represent positive and negative correlations, respectively.
Conclusion
We sequenced the genome of P. italicum GL_Gan1, which is a local strain in Guangzhou, China. A comparative genomics analysis among three Penicillium species, including P. italicum, P. digitatum, and P. expansum, suggested that the host range expansion from P. italicum and P. digitatum to P. expansum might be related to the expansion of protein families, genome restructuring, HGT, and positive selection pressure. In addition, we explored the molecular basis underlying the pathogenicity of P. italicum to Valencia Orange (Citrus sinensis). Diverse strategies mediating the pathogenicity of this fungus were revealed, including (1) the activation of effectors, such as CAZymes, SSCPs; (2) the suppression of host R genes; (3) the regulation of defense responses-related metabolites in host, such as trehalose and salicylic acid. However, in the present study, we have not compared diseased tissue to non-diseased tissue or fungal transcripts in pathogenic vs nonpathogenic interactions from a transcriptome perspective. More transcriptome analysis are necessary to elucidate the pathogen-host interaction. The data presented herein may be useful for further elucidating the molecular basis underlying the evolution of the host specificity of Penicillium species and for illustrating the the pathogenicity of P. italicum to Valencia Orange.
Materials and Methods
Fungal strain
Penicillium italicum GL_Gan1 was first isolated in Guangzhou, China from infected sweet orange exhibiting typical blue mold symptoms. The strain was purified as a monospore and stored at −80°C prior to use.
Fruit inoculation
The P. italicum GL_Gan1 conidiospores were suspended in sterile water containing 0.05% (v/v) Tween 80. The spore concentration in the suspension was adjusted to 1 × 107 spores/ml with a hemocytometer under an optical microscope (Olympus, Japan). Valencia orange (C. sinensis) was purchased from a local market and sterilized with 75% ethanol. Five cross-like wounds (width: 0.5–1; depth: 1–2 cm) were made at the base of the Valencia orange with sterilized tips. The wounds were then inoculated with 10 µl spore suspension, after which the fruits were incubated in a controlled-environment room (26 ± 1°C, natural photoperiod, and 70%–80% humidity). Infected Valencia orange peel tissues (flavedo and albedo) were collected at 0, 1, 3, 5, and 10 dpi, and then immediately frozen in liquid nitrogen for the subsequent RNA-seq and quantitative real-time PCR (qPCR) analyses. After inoculation, the tissues was immediately collected as the sample at 0 dpi.
Extraction of DNA and RNA
Penicillium italicum GL_Gan1 was cultured on potato dextrose agar for 7 days, after which spores were harvested. Genomic DNA was extracted from conidiospores (80 mg) with the HiPure Fungal DNA kit (Magen, Guangzhou, China). High-quality DNA was submitted to BGI-Shenzhen (Shenzhen, China) for genome sequencing. Total RNA was extracted from the inoculated Valencia orange peel tissue with TRIzol® reagent according to the manufacturer’s instructions (Invitrogen, China).
Genome sequencing and assembly
The P. italicum GL_Gan1 genomic DNA was used to construct 500-bp sequencing libraries. Sequencing data comprising 1,815 Mb (125-bp paired-end) were generated with the Illumina HiSeq™ 2000 system at BGI-Shenzhen (Shenzhen, China). To ensure the accuracy of the assembly, reads with 25 low-quality (≤ Q2) bases, 10% Ns, or a 15-bp overlap between the adapter, and duplicated reads were eliminated. The short reads were assembled with SOAPdenovo 1.05, with the assembly optimized with the key parameter K = 63.
Gene prediction and annotation
Homology-based and de novo methods were used to predict the genes in the assembled genome. For the homology-based prediction, genes encoding P. digitatum Pd1 proteins were mapped onto the assembled genome with Genewise [Citation51]. Regarding the de novo prediction, the Augustus program [Citation52] was used, with appropriate parameters. Data from these complementary analyses were merged to produce a non-redundant reference gene set using GLEAN (http://glean-gene.sourceforge.net/). Repeat sequences were identified with Repeat Masker (version 3.3.0) and Repbase (version 18.05), with the following parameters: – nolow – no_is – norna-s – engine wublast – parallel 1. Repeat Protein Mask was also used with the following parameters: – noLowSimple – pvalue = 1e-4 (http://www.repeatmasker.org/). Tandem repeats in DNA sequences were detected with Tandem Repeat Finder (version 4.0.4), with the following parameters: 2 7 7 80 10 50 Period_size – d – h seqfile (http://tandem.bu.edu/trf/trf.html). The protein-encoding genes were annotated via BLASTP searches of the SwissProt (release: 2012_06), GO (release: 1.419), COG (release: 20,090,331), KEGG (release: 59), and NR (20,150,222) databases, with a threshold e-value ≤ 1e-5.
The CAZyme database (http://www.cazy.org/) was used to identify proteins involved in carbohydrate metabolism. Pathogenicity- and virulence-related genes were identified using the PHI-base database (http://www.phibase.org/). We identified PKS and NRPS with SMURF [Citation53]. Cytochrome P450 s were identified with the Cytochrome P450 database (http://drnelson.uthsc.edu/CytochromeP450.html). The SSCPs of P. italicum GL_Gan1 were analyzed as described by Que et al. [Citation28]. The GPCR sequences were evaluated regarding their seven transmembrane regions with Phobius [Citation54] and the default settings of TMHMM 2.0. The R genes were identified as previously described [Citation44]. The STEM program (version 1.3.9) [Citation55] was used to compare and visualize the RNA-seq data.
Comparative genomics analysis
Protein sequences for P. italicum GL_Gan1, P. expansum CMP1, P. expansum Pd1, P. expansum MD8, P. italicum PHI1, P. digitatum PHI26, and A. nidulans FGSCA4 were aligned and redundant sequences were eliminated with BLAST and SOLAR (https://surfsara.nl/systems/lisa/software/solar). Genes were clustered with TreeFam and Hcluster_sg [Citation56], with -w 10 -s 0.34 -m 500 -b 0.1 -C category genes_file. After clustering, multiple protein sequences were aligned for each gene family with MUSCLE [Citation57], and the protein alignments were converted to CDS alignments.
The single-copy core genes of the above-mentioned six Penicillium species/strains and A. nidulans (outgroup species) were identified with Hcluster_sg [Citation56]. Multiple sequences were then aligned with MUSCLE [Citation57]. The phyml subprogram of TreeBeST (http://treesoft.sourceforge.net/treebest.shtml) was used to construct a phylogenetic tree, with default parameters and 1,000 bootstrap replicates. The core genome and pan-genome of the six Penicillium species/strains were obtained based on a previous analysis of a Yersinia pestis population [Citation58]. Additionally, Ka and Ks were estimated using an established method [Citation59]. Horizontal gene transfer was analyzed according to a published procedure [Citation60]. Protein sequences that were shorter than 150 amino acids were excluded. The BLASTP algorithm was used for similarity searches involving the bacterial and viral protein databases of NR (1e-5, identity > 40%). Moreover, phylogenetic trees with fewer than 10 species were excluded. Multiple sequences were then aligned with MUSCLE [Citation57] MEGA v6.06 (https://www.megasoftware.net/) was used to construct Neighbor-joining tree with default parameters and 2,000 bootstrap replicates. With which the iTOL v4.0 (https://itol.embl.de) was employed to improve the display, annotation and management of phylogenetic trees.
RNA sequencing
Two independent biological replicates of inoculated Valencia orange fruits were used to construct 10 short-fragment libraries with the Illumina TruSeq RNA library construction kit (version 2) (Illumina, CA, USA) for an RNA-seq analysis. After low-quality raw reads were discarded, the remaining high-quality reads were aligned to the P. italicum GL_Gan1 and Valencia orange gene sequences with SOAP2 [Citation61]. The RNA-seq data were analyzed and gene expression levels (as FPKM) were calculated with the RSEM software package [Citation62]. The NOIseq method was applied to screen for differentially expressed genes between two groups [Citation63]. An FDR ≤ 0.001 and an absolute value of the log2 ratio ≥ 1 were used as the criteria for identifying differentially expressed genes.
qPCR analysis
The PrimeScript™ RT reagent Kit with gDNA Eraser (Takara, Otsu, Japan) was used to synthesize cDNA. Gene-specific primer pairs (Table S5) were designed with the Primer Express software (Applied Biosystems, Foster City, CA, USA). G3PDH and Actin were used as reference gene. The qPCR analysis was completed with three biological replicates. The output data were generated with the Sequence Detector program (version 1.3.1) (Applied Biosystems) and then analyzed according to the 2−ΔΔCt method [Citation64].
Metabolomics analysis
Valencia orange peel tissues infected with P. italicum were collected at 3 dpi for a GC-TOF-MS analysis as described by Dunn et al. [Citation65]. Metabolites were extracted from 1.0 g citrus samples with a methanol:chloroform (3:1) solution, with 100 μl adonitol (0.2 mg/ml) as an internal standard. After derivatization, the extracts were added with fatty acid methyl esters (C8-C24) as retention index markers derivatized and then were transferred to a 7890 gas chromatograph system (Agilent Technologies, USA) connected to a Pegasus HT time-of-flight mass spectrometer (LECO Corporation, USA). Additionally, a DB-5 MS capillary column was coated with cross-linked 5% diphenyl/95% dimethyl polysiloxane (30 m × 250 μm inner diameter, 0.25 μm film thickness; J&W Scientific, Folsom, CA, USA). The Chroma TOF 4.3X software (LECO, St. Joseph, USA) and LECO-Fiehn Rtx5 database were used for the raw peak extraction, baselines correction, deconvolution analysis and peak identification. Both of retention time index (RT) and mass spectral similarity were considered in metabolites identification. If the similarity is >700, we deem the metabolite identification is reliable. If the similarity is < 200, the compound name is defined as “analyte”, and the similarity between 200 and 700, the compound name was considered as a putative annotation.
Principal component analysis (PCA) and orthogonal projections to latent structures-discriminant analysis (OPLS-DA) were used to display the similarity and difference of the origin data with the SIMCA-P 13.0 software package (Umetrics, Umea, Sweden).
To refine this analysis, the first principal component of variable importance projection (VIP) was obtained. The VIP values exceeding 1.0 were first selected as changed metabolites. In step 2, the remaining variables were then assessed by Student’s T test (T-test), P > 0.05, variables were discarded between two comparison groups.
Of the differentially accumulated metabolites, we quantified trehalose, salicylic acid, and D-sphingosine by UPLC-QQQ-MS. The standard chemicals were purchased from Sigma Chemical Co. (St. Louis, USA) and prepared as 100 µg/ml solutions. A 500-mg citrus peel sample was prepared and analyzed with the 6460 triple quadrupole LC/MS system with an electrospray ionization source (Torrance, CA, USA) as described by Chen et al. [Citation66]. Samples were examined with six repetitions. Additionally, the Pearson correlation coefficient was used to assess the relationship between the metabolite changes and gene expression levels. Furthermore, a heat map was prepared with the R program and the PathPod mapping system as described by Hsu et al. [Citation67].
Statistical analysis
Data are presented herein as the mean value of three biological replicates ± standard deviation. The significance of any differences among samples was calculated with SPSS (version 7.5) (SPSS, Inc., Chicago, IL, USA).
Data availability
The draft P. italicum GL_Gan1 genome sequence was deposited in the GenBank database (accession number LWEC00000000). The transcriptome sequence was deposited in the GenBank database (accession number SRP073474).
Supplemental Material
Download Zip (859.2 KB)Acknowledgments
This research was supported by National Key R&D Program of China (No. 2018YFD0401301), National Natural Science Foundation of China (No. 31772013), Science and Technology Planning Project of Guangzhou (Nos. 201710010135 and 201804020041). The wok also was supported by Guangdong Provincial Key Laboratory of Applied Botany, Key Laboratory of Plant Resource Conservation and Sustainable Utilization, CAS, and Key Laboratory of Post-Harvest Handling of Fruits, Ministry of Agriculture.
X.D. and L.G. designed and conceived the experiments. L.G., T.L., X.Z., X.C., M.H., and D.M. performed the experiments. Y. L., Y.X., J. Z., J.Y., and Q.Y. performed the sequencing and assembly of the genomes and bioinformatics analysis. M.H., B.T.H., R.J.H., and B.A.W. analyzed and interpreted the data. S.W., P.B., and B.A.W. provided reagents, materials, analytical tools, and support.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Jacques MA, Arlat M, Boulanger A, et al. Using ecology, physiology, and genomics to understand host specificity in Xanthomonas. Annu Rev Phytopathol. 2016;54(163–187).
- Stukenbrock EH. Evolution, selection and isolation: A genomic view of speciation in fungal plant pathogens. New Phytol. 2013;199(4):895–907.
- Friesen TL, Stukenbrock EH, Liu Z, et al. Emergence of a new disease as a result of interspecific virulence gene transfer. Nat Genet. 2006;38(8):953–956.
- Giraud T, Gladieux P, Gavrilets S. Linking the emergence of fungal plant diseases with ecological speciation. Trends Ecol Evol. 2010;25(7):387–395.
- Vilanova L, Vinas I, Torres R, et al. Infection capacities in the orange-pathogen relationship: Compatible (Penicillium digitatum) and incompatible (Penicillium expansum) interactions. Food Microbiol. 2012;29(1):56–66.
- Marcet-Houben M, Ballester AR, de la Fuente B, et al. Genome sequence of the necrotrophic fungus Penicillium digitatum, the main postharvest pathogen of citrus. BMC Genomics. 2012;13:646.
- Ballester A-R, Marcet-Houben M, Levin E, et al. Genome, transcriptome, and functional analyses of Penicillium expansum provide new insights Into secondary metabolism and pathogenicity. Mol Plant Microbe Interact. 2015;28(3):232–248.
- Li B, Zong Y, Du Z, et al. Genomic characterization reveals insights into patulin biosynthesis and pathogenicity in Penicillium species. Mol Plant Microbe Interact. 2015;28(6):635–647.
- Julca I, Droby S, Sela N, et al. Contrasting genomic diversity in two cosely related postharvest pathogens: Penicillium digitatum and Penicillium expansum. Genome Biol Evol. 2016;8(1):218–227.
- Marcet-Houben M, Gabaldon T. Horizontal acquisition of toxic alkaloid synthesis in a clade of plant associated fungi. Fungal Genet Biol. 2016;86:71–80.
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43(D1):D222–D226.
- Ma M, Wang Z, Li L, et al. Complete genome sequence of Paenibacillus mucilaginosus 3016, a bacterium functional as microbial fertilizer. J Bacteriol. 2012;194(10):2777–2778.
- Niehaus EM, Muensterkoetter M, Proctor RH, et al. Comparative “omics” of the fusarium fujikuroi species complex highlights differences in genetic potential and metabolite synthesis. Genome Biol Evol. 2016;8(11):3574–3599.
- Sanchez-Vallet A, Fouche S, Fudal I, et al. The genome biology of effector gene evolution in filamentous plant pathogens. Annu Rev Phytopathol. 2018;56(1):21–40.
- Lu S, Edwards MC. Genome-wide analysis of small secreted cysteine-rich proteins identifies candidate effector proteins potentially involved in Fusarium graminearum-wheat interactions. Phytopathology. 2016;106(2):166–176.
- Garrigues S, Gandia M, Castillo L, et al. Three antifungal proteins from Penicillium expansum: Different patterns of production and antifungal activity. Front Microbiol. 2018;9:2370.
- Looi HK, Toh YF, Yew SM, et al. Genomic insight into pathogenicity of dematiaceous fungus Corynespora cassiicola. Peer J. 2017;5:e2841.
- Satjarak A, Graham LE, Verbruggen H. Genome-wide analysis of carbohydrate-active enzymes Pyramimonas parkeae (Prasinophyceae). J Phycol. 2017;53(5):1072–1086.
- Bautista-Banos S. Postharvest Decay Control Strategies. Chapter 2 Penicillium digitatum, Penicillium italicum (Green Mold, Blue Mold); Elsevier, Academic Press; 2014, p.45–102.
- Banani H, Marcet-Houben M, Ballester AR, et al. Genome sequencing and secondary metabolism of the postharvest pathogen Penicillium griseofulvum. BMC Genomics. 2016;17(1):19.
- Yoshida K, Saunders DGO, Mitsuoka C, et al. Host specialization of the blast fungus Magnaporthe oryzae is associated with dynamic gain and loss of genes linked to transposable elements. BMC Genomics. 2016;17(1):370.
- Shen Q, Liu Y, Naqvi NI. Fungal effectors at the crossroads of phytohomone singaling. Curr Opin Microbiol. 2018;46:1–6.
- Lopez D, Ribeiro S, Label P, et al. Genome-wide analysis of Corynespora cassiicola leaf fall disease putative effectors. Front Microbiol. 2018;9:276.
- Moreira LRS, Filho EXF. An overview of mannan structure and mannan-degrading enzyme systems. Appl Microbiol Biotechnol. 2008;79(2):165–178.
- Del Carmen Rodriguez-Gacio M, Iglesias-Fernandez R, Carbonero P, et al. Softening-up mannan-rich cell walls. J Exp Bot. 2012;63(11):3975–3988.
- Baath JA, Giummarella N, Klaubauf S, et al. A glucuronoyl esterase from Acremonium alcalophilum cleaves native lignin-carbohydrate ester bonds. FEBS Lett. 2016;590(16):2611–2618.
- Kumamoto CA. Molecular mechanisms of mechanosensing and their roles in fungal contact sensing. Nat Rev Microbiol. 2008;6(9):667–673.
- Que Y, Xu L, Wu Q, et al. Genome sequencing of Sporisorium scitamineum provides insights into the pathogenic mechanisms of sugarcane smut. BMC Genomics. 2014;15(1):996.
- Dean RA, Talbot NJ, Ebbole DJ, et al. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature. 2005;434(7036):980–986.
- Goldstone JV, Goldstone HMH, Morrison AM, et al. Cytochrome p450 1 genes in early deuterostomes (tunicates and sea urchins) and vertebrates (chicken and frog): Origin and diversification of the CYP1 gene family. Mol Biol Evol. 2007;24(12):2619–2631.
- Fox EM, Howlett BJ. Secondary metabolism: regulation and role in fungal biology.Curr Opin Microbiol. [2008]; 11(6):481–487.
- Hu X, Xiao G, Zheng P, et al. Trajectory and genomic determinants of fungal-pathogen speciation and host adaptation. Proc Natl Acad Sci U S A. 2014;111(47):16796–16801.
- Ma LJ, van der Does HC, Borkovich KA, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464(7287):367–373.
- Wang S, Leclerque A, Pava-Ripoll M, et al. Comparative genomics using microarrays reveals divergence and loss of virulence-associated genes in host-specific strains of the insect pathogen Metarhizium anisopliae. Eukaryot Cell. 2009;8(6):888–898.
- Sharma R, Mishra B, Runge F, et al. Gene loss rather than gene gain Is associated with a host jump from monocots to dicots in the smut fungus Melanopsichium pennsylvanicum. Genome Biol Evol. 2014;6(8):2034–2049.
- Elbarbary RA, Lucas BA, Maquat LE. Retrotransposons as regulators of gene expression. Science. 2016;351(6274).
- Zhang L, Huang X, He C, et al. Novel fungal pathogenicity and leaf defense strategies are revealed by simultaneous transcriptome analysis of Colletotrichum fructicola and strawberry infected by this fungus. Front Plant Sci. 2018;9:434.
- Li P, Liu W, Zhang Y, et al. Fungal canker pathogens trigger carbon starvation by inhibiting carbon metabolism in poplar stems. Sci Rep. 2019;9(1):10111.
- Wu G, Ii WM J, Lichtner FJ, et al. Whole-genome comparisons of Penicillium spp. reveals secondary metabolic gene clusters and candidate genes associated with fungal aggressiveness during apple fruit decay. Peer J. 2019;7:e6170.
- Houterman PM, Cornelissen BJC, Rep M. Suppression of plant resistance gene-based immunity by a fungal effector. PLoS Pathog. 2008;4(5):e1000061.
- El Hadrami A, El-Bebany AF, Yao Z, et al. Plants versus fungi and oomycetes: pathogenesis, defense and counter-defense in the proteomics era. Int J Mol Sci. 2012;13(6):7237–7259.
- Bradshaw RE, Guo Y, Sim AD, et al. Genome-wide gene expression dynamics of the fungal pathogen Dothistroma septosporum throughout its infection cycle of the gymnosperm host Pinus radiata. Mol Plant Pathol. 2016;17(2):210–224.
- Alkan N, Friedlander G, Ment D, et al. Simultaneous transcriptome analysis of Colletotrichum gloeosporioides and tomato fruit pathosystem reveals novel fungal pathogenicity and fruit defense strategies. New Phytol. 2015;205(2):801–815.
- Zheng F, Wu H, Zhang R, et al. Molecular phylogeny and dynamic evolution of disease resistance genes in the legume family. BMC Genomics. 2016;17(1):402.
- Tayeh C, Randoux B, Vincent D, et al. Exogenous trehalose induces defenses in wheat before and during a biotic stress caused by powdery mildew. Phytopathology. 2014;104(3):293–305.
- Zhang H, Hong Y, Huang L, et al. Virus-induced gene silencing-based functional analyses revealed the involvement of several putative trehalose-6-phosphate synthase/phosphatase genes in disease resistance against Botrytis cinerea and Pseudomonas syringae pv. tomato DC3000 in tomato. Front Plant Sci. 2016;7:1176.
- Wingler A. The function of trehalose biosynthesis in plants. Phytochemistry. 2002;60(5):437–440.
- Sanchez-Rangel D, Rivas-San Vicente M, Eugenia de la Torre-hernandez M, et al. Deciphering the link between salicylic acid signaling and sphingolipid metabolism. Front Plant Sci. 2015;6:125.
- La Mantia J, Unda F, Douglas CJ, et al. Overexpression of AtGolS3 and CsRFS in poplar enhances ROS tolerance and represses defense response to leaf rust disease. Tree Physiol. 2018;38(3):457–470.
- Aksic MMF, Dabic DC, Gasic UM, et al. Polyphenolic profile of pear leaves with different resistance to pear psylla (Cacopsylla pyri). J Agric Food Chem. 2015;63(34):7476–7486.
- Birney E, Durbin R. Using GeneWise in the Drosophila annotation experiment. Genome Res. 2000;10(4):547–548.
- Stanke M, Keller O, Gunduz I, et al. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34(Web Server):W435–W439.
- Khaldi N, Seifuddin FT, Turner G, et al. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol. 2010;47(9):736–741.
- Kall L, Krogh A, Sonnhammer ELL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338(5):1027–1036.
- Ernst J, Bar-Joseph Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinformatics. 2006;7(1):191.
- Ruan J, Li H, Chen Z, et al. TreeFam: 2008 update. Nucleic Acids Res. 2008;36(Database):D735–D740.
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797.
- Cui Y, Yu C, Yan Y, et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc Natl Acad Sci U S A. 2013;110(2):577–582.
- Yang ZH, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000;17(1):32–43.
- Nikoh N, McCutcheon JP, Kudo T, et al. Bacterial genes in the aphid genome: Absence of functional gene transfer from Buchnera to its host. PLoS Genet. 2010;6(2):e1000827.
- Li R, Yu C, Li Y, et al. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–1967.
- Li B, Dewey CN. RSEM: Accurate transcript quantific4ation from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):323.
- Tarazona S, Furio-Tari P, Turra D, et al. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015;43(21):e140.
- Jiang G, Xiao L, Yan H, et al. Redox regulation of methionine in calmodulin affects the activity levels of senescence-related transcription factors in litchi. Biochim Biophys Acta Gen Subj. 2017;1861(5):1140–1151.
- Dunn WB, Broadhurst D, Begley P, et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc. 2011;6(7):1060–1083.
- Chen D, Lin S, Xu W, et al. Qualitative and quantitative analysis of the major constituents in shexiang tongxin dropping Pill by HPLC-Q-TOF-MS/MS and UPLC-QqQ-MS/MS. Molecules. 2015;20(10):18597–18619.
- Hsu HH, Araki M, Mochizuki M, et al. A systematic approach to timeseries metabolite profiling and RNA-seq analysis of Chinese hamster ovary cell culture. Sci Rep. 2017;7:43518.