
ABSTRACT
In 2014, enterovirus D68 (EV-D68) emerged causing outbreaks of severe respiratory disease in children worldwide. In a subset of patients, EV-D68 infection was associated with the development of central nervous system (CNS) complications, including acute flaccid myelitis (AFM). Since then, the number of reported outbreaks has risen biennially, which emphasizes the need to unravel the systemic pathogenesis in humans. We present here a comprehensive review on the different stages of the pathogenesis of EV-D68 infection – infection in the respiratory tract, systemic dissemination and infection of the CNS – based on observations in humans as well as experimental in vitro and in vivo studies. This review highlights the knowledge gaps on the mechanisms of systemic dissemination, routes of entry into the CNS and mechanisms to induce AFM or other CNS complications, as well as the role of virus and host factors in the pathogenesis of EV-D68.
Graphical abstract
Introduction
Enterovirus-D68 (EV-D68) is a non-enveloped, positive-sense single-stranded RNA virus that belongs to the family Picornaviridae, genus Enterovirus. Most picornaviruses, such as poliovirus, hepatitis A virus and coxsackievirus, are transmitted through the oral-fecal route. Other members, however, such as rhinovirus and EV-D68, are respiratory viruses and transmitted via the respiratory route. EV-D68 infection generally results in mild upper respiratory symptoms, but it can also cause severe lower respiratory tract disease, leading to hospitalization, intubation and occasionally death. To a great extent, children have an increased risk to develop severe diseases from EV-D68 infection, but reports have also shown that adults, especially those with underlying hematological malignancies or immune suppression, can also develop severe EV-D68-associated respiratory diseases [Citation1–3]. EV-D68 infection is also associated to a variety of central nervous system (CNS) complications, of which acute flaccid myelitis (AFM) is reported most commonly. The total number of EV-D68-associated AFM cases has been increasing during the biennially EV-D68 epidemics since 2014 [Citation4–7]. Clinically, EV-D68-associated AFM overlaps largely with acute flaccid paralysis (AFP) caused by poliovirus, which has caused large, documented outbreaks worldwide since 1940 until the development of an effective vaccine in 1962 [Citation8,Citation9]. However, some clinical features, including paralysis of upper limbs and cranial nerve dysfunction, are more frequently reported in AFM patients infected with EV-D68 rather than poliovirus [Citation10,Citation11].
The life cycle of EV-D68 is similar to that of other enteroviruses. Following attachment to a host cell, the virus is internalized through endocytosis. Viral RNA is transferred across the endosomal membrane into the cytoplasm, where replication takes place. Upon replication, assembly and virion maturation, newly produced viruses are released from the host cell through the release of extracellular vesicles or through the induction of cell lysis [Citation12].
To date, EV-D68 has evolved into four phylogenetic clades: A, B, C and D. Clade A is divided into subclades A1 and A2, although the latter is now considered a different clade and reclassified as clade D. Clade D diverged into subclades D1 [Citation13,Citation14] and D2 [Citation15]. Clade B is divided into subclades B1, B2 and B3. The different clades seem to co-circulate, although some clades are more prevalent than others. In 2014, all viruses isolated during the outbreak in the USA belonged to subclade B1 [Citation16]. However, after 2014, viruses from subclade B3 became more prominent [Citation17] and, to a lesser extent, D1 [Citation13]. Although it was initially believed that the ability to cause CNS complications was clade-specific, studies have now shown that clade B3 isolates from the 2014 outbreak differ in their ability to cause CNS complications in vivo and replication efficiency in vitro, suggesting that the ability to invade and replicate in the CNS is not a clade-specific feature [Citation18–20].
Unlike poliovirus, there is no vaccine available for EV-D68, despite its ability to cause neurological symptoms and acute paralysis in children. Moreover, because EV-D68 cases were only reported sporadically before 2014, and diagnostics for EV-D68 are not a standard practice in most laboratories, the burden of disease and trends are not fully understood. In order to be able to develop preventive or intervention measures, knowledge on the pathogenesis of the virus infection is essential. In this review, we present a concise overview on the current knowledge on the different stages of the pathogenesis of EV-D68 infections: respiratory infection, systemic dissemination and CNS invasion
EV-D68 receptors and their distribution
The tissue and cell tropism of a virus are determined by many factors, of which cellular receptors on host cell surfaces are considered to be an important one. Several studies have identified functional EV-D68 receptors in vitro, but the association of these receptors with the tropism of EV-D68 in vivo has not been studied comprehensively. Both α2,6-linked sialic acids (SAs) and α2,3-linked SAs serve as an essential functional receptor for EV-D68 [Citation21,Citation22]. The prototype laboratory-adapted EV-D68 strain Fermon has a stronger affinity to α2,6-linked SAs than α2,3-linked SAs in vitro [Citation22]. Whether current circulating clinical isolates of EV-D68 have a preference toward α2,6-linked SAs or α2,3-linked requires further investigation. In humans, α2,6-linked SAs are abundantly expressed on ciliated epithelial cells of the upper respiratory tract (URT; nasal cavity, nasopharynx and oropharynx) and lower respiratory tract (LRT; trachea, bronchi and bronchioles). In the alveoli, α2,6-linked SAs are predominantly expressed on type I pneumocytes. In contrast, α2,3-linked SAs are predominantly expressed in the LRT on club cells in the bronchioles and type II pneumocytes in the alveoli [Citation23,Citation24] (). Human lung epithelial cell lines, including A549, express SAs, which corresponds to the susceptibility of these cells to EV-D68 infection in vitro [Citation25,Citation26]. Haploid genetic screening revealed that infection of the laboratory-adapted Fermon strain depends on SA, whereas some clinical isolates from 2009 and 2010 from the Netherlands are SA-independent suggesting a potential role for other functional receptors [Citation21]. Sialic acids are also abundantly expressed beyond the respiratory tract but this has not been studied in the context of tissue or cell tropism for EV-D68. In vitro, infection of neuroblastoma (SK-N-SH) cells with EV-D68 isolates from 2009 to 2016 from the Netherlands is in part associated with attachment to SAs [Citation20].
Table 1. Cellular receptors for EV-D68 and their expression on different cells types of the respiratory tract, CNS and lymphoid system SAs: Sialic acids; CNS: central nervous system; DCs: dendritic cells; NA: not available; *: in vitro study
In addition to SAs, heparan sulfate proteoglycans have been identified as a functional receptor for EV-D68 in vitro [Citation27]. Whether heparan sulfate is also a functional receptor in vivo remains to be determined, since it is associated with cell culture-adaptive mutations [Citation20,Citation27]. In human airway epithelial cells, heparan sulfate is mainly expressed on the basolateral side, which suggest that heparan sulfate might not be an important receptor in the human respiratory tract. However, heparan sulfate is abundantly expressed on cells outside the respiratory tract, such as cells of the CNS and muscle cells [Citation28–30] (), and could therefore contribute to EV-D68 infection outside the respiratory tract.
Intercellular adhesion molecule 5 (ICAM-5) has been identified as a functional receptor for SA- dependent and -independent EV-D68 isolates in vitro [Citation31]. ICAM-5 is expressed on human rhabdomyosarcoma (RD), human embryonic kidney (HEK) 293 T and HeLa cells [Citation31,Citation32]. ICAM-5 expression in vivo is restricted to the somatodendritic membrane of neurons of the telencephalon in the brain, so ICAM-5 is not expressed on cells in the brainstem and spinal cord [Citation32,Citation33]. All known cellular receptors for EV-D68 and their presence in various cell types of the respiratory tract, CNS and lymphoid system are summarized in .
Overall, EV-D68 can use different receptors to enter the host cell, and their expression varies among tissues. This is likely an important factor for the broad tissue and cell tropism of EV-D68 in mammals, including respiratory epithelial cells, muscle cells, lymphoid cells and even cells in the CNS. Even though differences in receptor recognition have been reported among EV-D68 isolates, it remains to be determined whether this has an impact on the tissue and cell tropism in vivo.
EV-D68 infection in the respiratory tract
Clinical observations
EV-D68 infection causes mild to severe respiratory diseases, especially in young children with a median age of 4 years old [Citation34,Citation35]. It is thought that during a mild or subclinical infection the virus mainly infects the URT, resulting in fever and coughing. However, more severe respiratory disease is observed when virus spreads to the LRT, resulting in dyspnea, wheezing pneumonia and hypoxia, and occasionally leads to the need for ventilator support and intensive care admission [Citation36]. Previous history of asthma or recurrent wheezing appears to be an important risk factor for the development of asthma exacerbation or pneumonia [Citation34,Citation37,Citation38]. However, severe pneumonia caused by EV-D68 has also been observed in patients without underlying diseases [Citation35].
Although EV-D68 infections are predominantly reported in children, viral RNA has been detected in adults that developed mild respiratory disease. This adult group included healthy adults [Citation13,Citation39,Citation40] as well as adults who were immunocompromised or had underlying co-morbidities, including hematologic malignancies or hematopoietic cell transplant patients [Citation1–3].
Pathogenesis
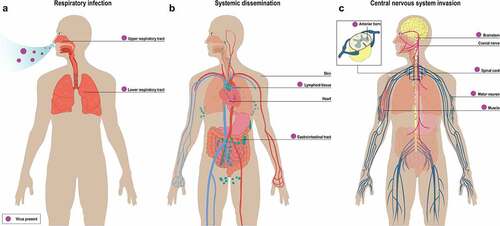
The pathogenesis of EV-D68 infection in the respiratory tract in humans is poorly studied. Few clinical studies have reported interstitial pneumonia with diffused and patchy alveolar infiltration in the lungs [Citation40,Citation41]. The fact that EV-D68 was detected in diagnostic samples from the URT and LRT suggests that the virus is able to replicate throughout the respiratory tract () [Citation1,Citation35]. Although receptors for EV-D68 are present in both human URT and LRT (), the exact cell tropism during mild or severe respiratory disease in humans is unknown.
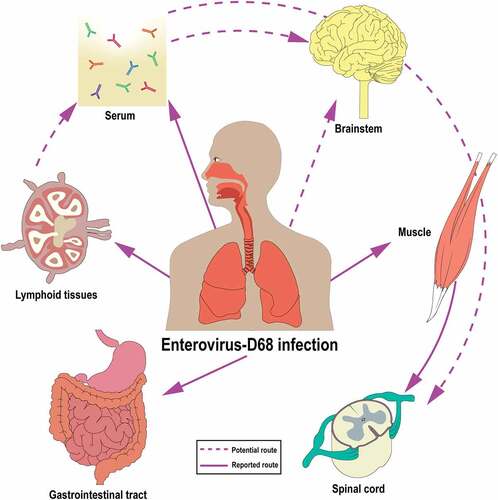
Figure 1. Hypothetical model of pathogenesis of EV-D68 infection based on findings from EV-D68 infected patients and experimental in vivo and in vitro studies. (a) EV-D68 infection in the respiratory tract: Inhaled virus particles firstly replicate in the upper respiratory tract and might spread to the lower respiratory tract. (b) EV-D68 infection outside the respiratory tract: Systemic dissemination of virus into the bloodstream and/or draining lymph nodes, resulting in viremia and infection of extra-respiratory tissues including the gastrointestinal tract, skin and heart. (c) EV-D68 infection in the CNS: EV-D68 may invade the CNS through infection of the muscles and subsequent spread via the neuromuscular junction to motor neurons in the anterior horn of the spinal cord using retrograde axonal transport. Other possible invasion mechanisms into the CNS are via the blood-brain barrier, blood-CSF barrier or cranial nerves. Purple dot represents EV-D68. Purple arrow represents the flow of EV-D68 systemic dissemination in the circulation
The cell tropism of EV-D68 in the URT has been studied using in vitro and in vivo models. In vitro, EV-D68 infected predominantly ciliated epithelial cells in human nasal airway cultures, causing cell lysis. The infection also resulted in the induction of several cytokines, such as interleukin (IL-8), IP-10, interferon- (IFN-) λ1/λ3, IL-1β, IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF) at 4 days post-inoculation (dpi) [Citation42]. In vivo, infectious virus and viral RNA were detected in nasal washes from intranasally inoculated ferrets (Mustela putorius furo) and cotton rats (Sigmodon hispidus), indicating virus replication in the URT [Citation43,Citation44]. Unfortunately, the cell tropism of EV-D68 and the associated inflammatory response in the URT have not been studied in these in vivo models.
The pathogenesis of LRT disease caused by EV-D68 has been studied to a limited extend in animal models. From cotton rats that were intranasally inoculated with viruses isolates from the USA in 2012 and 2014 or Fermon strain, infectious virus has been isolated from bronchoalveolar lavage (BAL) and lung tissue. Virus titers peaked at 24 hours post-inoculation (hpi) and rapidly dropped afterward. Rapid antiviral responses in the LRT were detected within 4 to 48 hpi, including inflammatory cytokines (IL-6 and IFN-γ), chemokines (GRO, MCP-1, IP-10 and RANTES), IFN-β and two IFN-inducible proteins (Mx-1 and Mx-2). Pathology showed mild peribronchiolitis and interstitial pneumonia at 48 hpi. Unfortunately, the tissue and cell tropism – by detection of virus antigen in vivo – was not determined [Citation43]. In ferrets intranasally inoculated with Fermon, viral RNA was detected in lung tissues 3 dpi. Acute inflammatory responses in the lungs included IL-1, IL-5 and IL-8 at 7 and 14 dpi. Histological evidence for mild interstitial pneumonia was observed from 3 to 7 dpi without any lesions in the trachea. Additionally, EV-D68 viral capsid protein VP1 was detected in the connective tissues surrounding the alveoli, which was associated with thickening of the alveolar wall. Double stainings for viral VP1 and cellular SAs in the ferret lungs revealed that EV-D68 exclusively infected cells that expressed α2,6-linked SAs [Citation44].
In vivo studies using mice – both BALB/c and IFN-α/β and -γ receptor-deficient mice – revealed that EV-D68 replicates within the lungs and peaks within the first 24 hpi [Citation45,Citation46]. In IFN-α/β and -γ receptor-deficient mice, increased levels of inflammatory cytokines and chemokines were detected in the lungs at 24 hpi, which included MCP-1 and IL-6, compared to sham-infected mice. Histopathological changes comprised of moderate interstitial pneumonia characterized by mononuclear cell infiltrates in the perivascular space and alveolar walls. In addition, EV-D68 viral capsid protein VP2 was detected at 48 hpi in alveolar epithelial cells [Citation45]. Infection of BALB/c mice with clinical isolates from 2014 resulted in elevated lung inflammatory cytokines and chemokines, including IL-17A and its target proteins CXCL1 and CXCL2, compared to sham-infected mice and rhinovirus-inoculated mice. EV-D68 viral capsid protein VP3 was detected in the lungs at 24 hpi. The number of neutrophils increased in the BAL fluid collected from these mice at 48 hpi . These findings suggest that EV-D68 induces neutrophilic airway inflammation facilitated by IL-17A, which may in part explain the development of asthma exacerbation following EV-D68 infection in humans [Citation46].
The studies described above suggest that EV-D68 is able to infect and replicate in the URT and LRT. In most individuals, infection is followed by the induction of innate immune responses, which limit virus replication and associated inflammation early after infection [Citation13,Citation40]. The mechanisms by which some individuals, especially children and immunocompromised adults, develop more severe disease are not fully understood [Citation1]. However, it is likely that host immune responses, or the lack thereof, is an important determinant, together with the different mechanisms of EV-D68 for evading the host innate immune responses. The latter have mainly been studied in vitro, but it is important to study the virus-host interaction and the associated immune response in more details in vivo [Citation47–49].
Altogether, to get more insight into the pathogenesis of EV-D68-induced respiratory disease, an animal model that mimics disease in humans is essential. Experimental infections in ferrets and cotton rats suggested that these animal models can be promising tools to study the pathogenesis of respiratory disease caused by EV-D68. However, the limited use of relevant EV-D68 clinical isolates and instead the use of laboratory-adapted virus strains might not fully represent the clinical disease in humans. Therefore, ideally future in vivo studies should use contemporary strains to unravel the receptor usage and associated cell tropism, as well as the host and viral factors that impact immune response and associated pathogenesis.
Systemic dissemination of EV-D68
EV-D68 can be detected outside the respiratory tract, although the frequency is not well known. Viral RNA has been detected in blood, sera and stool samples from EV-D68 infected patients and in cerebrospinal fluid (CSF) from EV-D68-associated AFM patients [Citation6,Citation7,Citation16,Citation39,Citation50,Citation51]. The mechanism of extra-respiratory spread is not completely understood, but will be discussed in this section.
Even though virus or viral RNA has been detected in the blood or serum of EV-D68 infected patients, the temporal kinetics of an viremia or RNAemia and whether this only occurs during severe disease are unclear. Unfortunately, from most patients, only respiratory tract samples are tested for EV-D68 (). In animal models, systemic spread of EV-D68 including viremia and RNAemia have been detected. Infectious virus particles have been isolated from lungs, blood, muscle, spinal cord, liver, kidneys and spleen of intranasally inoculated interferon-deficient four-week-old mice [Citation45,Citation52] and, similarly, viral RNAs were also detected in several tissues of intraperitoneally inoculated one-day-old mice [Citation53]. Viral RNAs were also been detected in the blood of ferrets intranasally inoculated with Fermon at 3 dpi and Chinese rhesus macaques (Macaca mulatta) inoculated with Kunming strain at 9 days post-inoculation [Citation44,Citation54].
Table 2. The detection of EV-D68 in diagnostic samples including whole blood, serum or plasma, stool and CSF samples in reported cases. CSF: cerebrospinal fluid; NA: not available. *: More than 1 sample were collected from a patient
The origin of virus in the blood is unknown but it is likely that infected lymphoid cells and lymphoid tissues contribute to a viremia. Other picornaviruses, such as poliovirus, have been found to replicate in CD11c+ macrophages or dendritic cells in the tonsil follicles [Citation55], but whether EV-D68 is capable to replicate in these or other immune cells in vivo remains to be determined. In vitro inoculation of human leukocytic cell lines with the Fermon strain have shown a productive infection of granulocytic (KG-1), monocytic (U-937), T (Jurkat and MOLT) and B (Raji) cell lines [Citation56]. Unfortunately, these studies have not been performed with clinical isolates. Nonetheless, this finding suggests that infection of leukocytes may play a role in EV-D68 viremia and thereby contribute to the systemic spread of EV-D68 in vivo. In vivo, viral RNA has been detected in the axillary lymph nodes of ferrets inoculated intranasally with the Fermon strain at 5 and 7 dpi [Citation44]. Intramuscular inoculation of cotton rats with a clinical isolate showed the presence of negative stranded viral RNA, indicative for virus replication, in the draining lymph nodes of the site of inoculation [Citation43].
EV-D68 could enter the bloodstream through the disruption of the respiratory epithelial-endothelial barrier, via basolateral release of virus from respiratory epithelial cells or through direct infection of pulmonary endothelial cells. EV-D68 infection of primary human nasal epithelial cells grown in air-liquid interface led to high cytotoxicity, as measured by lactate dehydrogenase release into the basal medium, and loss of tissue integrity, as shown by a decrease in transepithelial electrical resistance [Citation42]. Whether this disruption of epithelial integrity leads to spillover of virus particles into the bloodstream in vivo remains to be determined. Whether, EV-D68 infection of polarized epithelial cells leads to polarized release of new virus progeny, like observed with other respiratory viruses, is unknown. Newly produced influenza A viruses, for example, are released from the apical side of epithelial cells [Citation57]. Since EV-D68 infection leads to cell lysis at late stages of infection, it might be challenging to determine if there is basolateral release of viral progeny prior to cell lysis using in vitro models. Nonetheless, studies have shown that enteroviruses can exit the infected host cell in autophagosomes before cell lysis and EV-D68 has also been suggested to reshape the autophagic pathway to promote virus replication and egress [Citation12,Citation58]. Whether EV-D68 infection of respiratory epithelial cells results in basolateral release of these autophagosome-like vesicles containing new virus progeny remains to be determined. Infection of endothelial cells by EV-D68 has not been reported. Other picornaviruses have been shown to infect various endothelial cells in vitro, but whether this occurs in vivo is unknown [Citation59–66].
EV-D68-associated acute gastroenteritis has been reported, suggesting that the virus, despite being a respiratory virus, may also be able to infect the gastrointestinal tract like other enteroviruses [Citation67]. This enterotropism is supported by the fact that EV-D68 is occasionally detected in stool samples from patients () [Citation50,Citation68–70]. EV-D68 has also been shown in vitro to infect human intestinal epithelial cell lines. Inoculation of human colorectal adenocarcinoma HT-29 and LS174T cell lines with the Fermon strain resulted in infection, based on the detection of viral RNA and intracellular VP1, but not in efficient replication, based on the low RNA levels and virus titers in the supernatant [Citation71]. Ferrets inoculated intranasally with the Fermon strain showed that viral RNA could be detected in feces at 3 dpi [Citation44]. Similar observation were observed in Chinese rhesus macaques inoculated intranasally with the Kunming strain, of which viral RNA peaked at 3 dpi in feces [Citation54]. The rare detection of EV-D68 in stool samples of infected patients, the inefficient replication efficiency in vitro, together with the early and transient detection of EV-D68 in stool samples of experimentally infected ferrets or macaques suggest that EV-D68 replication in the gastrointestinal tract is suboptimal.
Other extra-respiratory complications associated with EV-D68 infection have also been reported. Some patients with EV-D68 infection developed cardiac symptoms, such as myocarditis, pericarditis and acute cardiac failure [Citation72]. Several clinical cases have reported the appearance of skin rash in EV-D68 patients [Citation73–75]. Whether the virus infects the cells of the heart, which leads to cardiac symptoms, or the dermis and epidermis, which leads to skin rash, remains to be determined. Intranasal or intraperitoneal inoculation of mice resulted in infection of limb muscles and the development of muscle disease, suggesting that EV-D68 is capable of replicating in muscle cells in vivo [Citation45,Citation76]. However, whether muscle cells are infected in humans is still unknown. Potential replication sites and affected tissues during the systemic dissemination of EV-D68 are summarized and illustrated in .
All in all, the frequency and mechanism of the systemic dissemination of EV-D68 remains poorly understood. However, as systemic spread of EV-D68 is an important factor in the pathogenesis of EV-D68, it is important to get more insight into the frequency and temporal kinetics of systemic spread as well as its association with the development of severe disease and CNS complications. In addition, future studies should reveal if systemic spread is solely a spillover of virus produced in the respiratory tract, or if virus is transported – either cell-free or cell-associated – to lymphoid tissues where virus is amplified and released into the circulation.
EV-D68 infection in the central nervous system
Clinical observations
Various CNS complications are associated with EV-D68 infections, of which the development of AFM is being reported most frequently. In addition, EV-D68 infection has been associated with cranial nerve dysfunction, encephalitis, meningoencephalitis and aseptic meningitis. Signs of CNS complications mostly occur following febrile upper respiratory tract symptoms, such as rhinorrhea, coughing and pharyngitis, and gastrointestinal symptoms, such as vomiting and diarrhea. EV-D68-associated CNS disease is challenging to diagnose since the virus is rarely detected in CSF (). However, EV-D68 viral RNA can be detected in nasopharyngeal samples in the first week after onset of CNS complications, after which the chance to detect of EV-D68 in respiratory samples declines [Citation10,Citation68,Citation77,Citation78].
The development of EV-D68-associated AFM can progress rapidly, even within hours to days after onset of symptoms [Citation68,Citation79]. Early manifestations of AFM include headache, stiffness or pain in the back and neck or affected limb, followed by reduced or absent reflexes [Citation10,Citation68]. One to four limbs can be affected with an asymmetric distribution. Generally, upper limbs are more commonly affected than lower limbs [Citation10,Citation68,Citation77,Citation78]. In severe cases, the diaphragm muscles may also be affected, resulting in the need for ventilator support [Citation78,Citation80]. Magnetic resonance imaging (MRI) showed lesions in the anterior horn of the spinal cord, especially in cervical regions (C2-C8) and upper thoracic regions (T1-T3). In addition, electrodiagnostic studies showed a motor neuropathy at C5-C8 level [Citation78]. Together, these clinical and diagnostic evidence support that EV-D68 can impair motor neurons in the spinal cord. Furthermore, nearly all patients had notable muscle atrophy in the affected limbs within weeks to months after paralysis onset [Citation10] and diffused muscle aches and limb pain that lasted up to weeks or months after onset [Citation68].
Other neurological complications have also been associated with EV-D68 infections [Citation68,Citation77]. Cranial nerve dysfunction has been observed in AFM patients, which correlated with brainstem lesions in cranial motor nuclei within the pons, medulla and midbrain by MRI [Citation78,Citation81]. Interestingly, a number of patients had cranial nerve dysfunction without clinical evidence for AFM [Citation10,Citation79]. Cranial nerve dysfunction was mostly characterized by facial weakness associated with the facial nerve (cranial nerve VII), diplopia associated with the abducens nerve (cranial nerve VI), or hypophonia, dysarthria and dysphagia associated with the glossopharyngeal nerve (cranial nerve IX) and the vagus nerve (cranial nerve X) [Citation10,Citation77]. Furthermore, some patients experienced sensory deficit in paralyzed limbs or autonomic deficit associated with bowel and/or bladder dysfunction [Citation68,Citation79].
EV-D68 infection is occasionally associated with encephalitis, meningoencephalitis or aseptic meningitis. Brainstem encephalitis was reported in an EV-D68-infected child and led to cardiopulmonary failure [Citation82]. A fatal meningoencephalitis was reported in a previously healthy child with EV-D68-associated AFM and severe pneumonia [Citation7]. Non-fatal aseptic meningitis has been reported in both children and adults [Citation2,Citation6,Citation16,Citation83–85]. Interestingly, the presence of EV-D68 RNA in the CSF seems to be more often detected in patients with aseptic meningitis than in AFM patients, although this may partly due to underreporting [Citation83].
Pathogenesis
The unprecedented emergence of CNS complications associated with EV-D68 has raised several questions about how the virus enters and causes diseases in the CNS. Mechanisms of virus entry and subsequent virus spread within the CNS and associated immune response in vivo remains largely unknown. Lesions in the anterior horn of the spinal cord are associated with AFM, suggesting that motor neuron function is impaired during the pathogenesis of EV-D68-associated AFM [Citation10,Citation78]. Furthermore, lesions in the cranial motor nuclei of the pons, medulla and midbrain are associated with cranial nerve dysfunction [Citation81,Citation86]. Whether these lesions are a direct result of virus infection needs to be confirmed, but the detection of EV-D68 RNA in the CSF of someAFM patients suggests that virus can invade the CNS [Citation2,Citation7,Citation16,Citation83,Citation86]. To better understand the pathogenesis of EV-D68 infections in the CNS, we will discuss possible routes for neuroinvasion and subsequent virus replication within the CNS and associated inflammatory responses.
Neuroinvasion
In general, viruses can gain access into the CNS via peripheral nerves or the hematogenous route. Virus entry into the CNS via peripheral nerves requires axonal transport. Mice inoculated intramuscularly with EV-D68 developed paralysis associated with motor neuron loss within 3 to 4 dpi [Citation32]. The same study also showed that EV-D68 was transported retrograde, but not anterograde, using axonal microtubules in motor neurons derived from human induced pluripotent stem cells (hiPSCs) [Citation32]. In vivo evidence for virus spread to the CNS via peripheral nerves are mainly derived from mice studies. Intraperitoneal inoculation of newborn mice or IFN-α/β/γ receptor-deficient mice with recent USA isolates of EV-D68 resulted in infection of muscle cells of the limb, followed by detection of virus in the spinal cord [Citation18,Citation53,Citation76,Citation87]. This suggests that EV-D68 virus can enter motor neurons via the neuromuscular junction, which is the direct connection between the muscle cells and the motor neurons. However, replication in muscle cells detected in mice after intranasal infection with a clinical isolate from 2014 or mouse adapted EV-D68 isolates did not always lead to neuroinvasion [Citation45,Citation76].
Cranial nerve dysfunction has been observed in a number of patients without limb paralysis [Citation10]. Since affected cranial nerves, like the facial, abducens, glossopharyngeal and vagus nerves innervate muscles of the face, respiratory tract, oral cavity and tongue with motor neurons or directly innervate the respiratory mucosa and oral cavity with sensory neurons, EV-D68 might use these to enter the CNS. Whether EV-D68 can enter the CNS via cranial nerves and what the exact mechanism is require further investigation.
To date, there is no direct evidence that EV-D68 invades the CNS through the blood-brain-barrier (BBB) or blood-CSF barrier. Since EV-D68 can be detected in the blood circulation, the virus could possibly invade the CNS and spread across the BBB or blood-CSF barrier. Other viruses, such as poliovirus, have been suggested to cause leakage of the BBB through direct infection of brain microvascular endothelial cells in vitro and in vivo [Citation63,Citation88]. Another possible route for EV-D68 to enter the CNS is via infection of lymphocytes or other immune cells that enter the CNS. Nonetheless, the mechanism of CNS invasion of EV-D68 in vivo requires more in-depth studies as various routes of entry may result in the diversity of clinical CNS diseases. Possible replication sites and dissemination route in the CNS are summarized and illustrated in .
Taken together, infection of EV-D68 leads to a wide spectrum of clinical CNS complications, ranging from AFM to cranial nerve dysfunction and meningoencephalitis. Even though the exact mechanism of CNS invasion is not known, it is possible that different invasion strategies used by the virus can explain the different disease manifestations.
Replication in the CNS
The cell tropism of EV-D68 in the CNS is not known, but postmortem pathological analyses has given some insights. In a patient with fatal meningomyeloencephalitis, extensive lymphocytic meningomyelitis and encephalitis were associated with neuronophagia in motor nuclei in meninges, cerebellum, midbrain, pons, medulla and cervical cord. In the spinal cord infiltrating CD3+ T cells were observed around motor nuclei and infiltrating CD20+ B cells in perivascular areas [Citation7]. Detection of EV-D68 proteins or viral RNA was not included making it challenging to link histological findings directly with local virus replication, even though EV-D68 RNA was detected in the CSF. In other EV-D68 patients, brainstem lesions were also observed by MRI although these were not further investigated [Citation10,Citation78,Citation81].
In vivo, viral RNA and virus antigen have been detected in the spinal cord of intraperitoneally and intracranially inoculated mice [Citation18,Citation53,Citation87]. Intracranial inoculation of mice with clinical isolates from 2014 or a prototype strain of EV-D68 resulted in minimal to absent virus replication in the cerebrum and cerebellum but efficient replication in motor neurons in the anterior horn of the spinal cord. This suggest that EV-D68 viruses can spread through the CNS toward the motor neurons in the spinal cord, but the mechanism remains unclear [Citation18].
The current understanding on the cell tropism of EV-D68 in the CNS relies in part on in vitro studies, which do not always correspond to in vivo observations. In vitro, human (SH-SY5Y and SK-N-SH) and mouse neuroblastoma cell lines [Citation19,Citation20,Citation26], together with motor neurons, cortical neurons and astrocytes derived from hiPSCs are permissive for EV-D68 infection [Citation19,Citation32]. Motor neurons derived from hiPSCs supported virus replication for 72 hours without a cytopathic effect, which contrasts to the death of motor neurons in intramuscularly inoculated mice [Citation26]. Moreover, in vivo, virus replication within the CNS of intracranially inoculated mice was restricted to motor neurons of the spinal cord [Citation18], while in vitro data based on hiPSCs showed permissiveness of human cortical neurons and astrocytes [Citation19]. In ex vivo organotypic brain slices of neonatal mice EV-D68 isolates from 2014 replicated efficiently, but the phenotype of EV-D68-infected cells in this model was not determined [Citation19].
The functional receptor or receptors of EV-D68 in the CNS are not known. Both sialic acids and ICAM-5 could facilitate virus entry into neuronal cells [Citation31,Citation32]. However, ICAM-5 is not expressed on motor neurons [Citation32] (). Another possible receptor that may play a role in the cell tropism of EV-D68 is heparan sulfate, since it is abundantly expressed on cells of the CNS [Citation28]. These observations suggest that EV-D68 could possibly infect and replicate in neurons and astrocytes. Whether this also occurs in vivo and whether the cell tropism contributes to differences in the clinical presentation requires further investigations.
Taken together, data from humans, in vivo mice models and in vitro studies suggest that EV-D68 has a preference for motor neurons while the susceptibility of other cells in the CNS varies among studies. Whether infection of motor neurons is necessary to cause paralysis remains unclear. In vivo studies in mice have shown that infection of muscle tissues without lesions in the spinal cord or motor neuron depletion resulted in paralysis [Citation76]. It is plausible that paralysis and muscle atrophy, which are commonly observed within weeks to months after weakness onset, are a result of viral-induced myositis, destruction of motor neurons, or a combination of both. To get more insight into the pathogenesis of EV-D68-associated paralysis future in vivo studies should focus on virus induced damage muscles tissues, including analysis of muscle enzymes in the blood as well as the detection of viral RNA and antigen in muscle tissues during the course of infection. In conclusion, the cell tropism of EV-D68 in cells of the CNS and how this is associated with clinical outcomes remains to be determined.
Viral factors important for the pathogenesis of EV-D68
Even though different clades of EV-D68 have circulated globally over time, the role of viral factors in the pathogenesis of EV-D68-associated respiratory and neurological diseases remains unknown. During the reemergence in 2014, multiple clades co-circulated but subclade B1 was predominant and associated with the development of severe respiratory diseases and AFM [Citation16]. After 2014, AFM has been reported in patients infected with different clades, including subclades B1, B3 and D1 [Citation13,Citation89,Citation90]. Whether genetic changes are responsible for the increased number of patients with severe respiratory disease and occurrence of neurological diseases since 2014 remains unclear [Citation16,Citation91]. Sequence analyses of subclade B1 isolates from the outbreak in 2014 revealed 12 specific amino acid substitutions in both structural and non-structural viral proteins compared those of clades A, C, subclade B3 and Fermon strains. These amino acid substitutions were found in the 5ʹUTR as well as different viral proteins, including VP2, VP3, 2A, 2B, 2 C and 3D. Some of these amino acids are also observed in neurotropic enteroviruses, e.g. poliovirus, EV-A71 and EV-D70 [Citation91], but whether these amino acid substitutions are associated with an increased pathogenicity in vivo remains to be elucidated.
Phenotypic characterization of different EV-D68 isolates from different clades has been performed after 2014 to identify possible viral factors important for the pathogenicity of EV-D68. Structure and sequencing analyses of clade B isolates from 2014 have shown high genetic variation in VP1 [Citation92]. Despite this high genetic variation, in vitro and in vivo studies did not reveal differences in replication efficiency between strains among the different clades or derived before and after 2014 [Citation19,Citation20]. Nonetheless, one study revealed that EV-D68 isolates from 2014 were able to replicate in neuroblastoma SH-SY5Y cells but not isolates from before 2014 [Citation26]. However, amino acid substitutions are easily acquired in clinical EV-D68 isolates while passaging in cell culture, resulting in the recognition of heparan sulfate as an additional receptor for EV-D68. The recognition of heparan sulfate was associated with large phenotypic changes in vitro, which emphasizes the necessity to sequence virus stocks to maintain phenotypic characteristics of clinical isolates [Citation20].
EV-D68 nonstructural proteins 3 C and 2A have been shown to play a role in the suppression of innate immune responses in vitro [Citation47–49]. The 3 C protein supresses type I IFNs through cleavage of IFN regulatory factor 7 (IRF7) [Citation47] or through binding to melanoma differentiation-associated gene 5 (MDA5), consequently inhibiting its interaction with downstream adaptor protein MAVS [Citation48]. The 2A protein also plays a role in the viral immune evasion by cleaving tumor necrosis factor receptor-associated factor 3 (TRAF3) [Citation49]. Whether different EV-D68 strains utilize different strategies to evade innate immune responses and whether these strategies differ in different cell types is currently unknown.
Overall, how the evolution of EV-D68 clades in past years is associated with the cell tropism and pathogenecity of EV-D68 remains undetermined. Also, the possible role of individual viral proteins in the reemergence of EV-D68 remains unclear. Therefore, further studies should provide more insight into phenotypic differences among the different clades circulating, and how these influence the pathogenesis as well as the role of individual viral proteins.
Future perspectives
Despite its long circulation in the global population, the pathogenesis of EV-D68-associated respiratory,systemic and CNS diseases remains largely unknown. The ability of EV-D68 to be transmitted through inhalation and to cause mild respiratory diseases like rhinovirus may render this virus to appear less harmless than it actually is. EV-D68 is a serious global health threat, especially because its ability to cause CNS diseases comparable to that of poliovirus. Unlike poliovirus, there is no vaccine available to prevent EV-D68 associated disease. To date, the numbers of reported infections and mortality cases were not as high as those for poliovirus. However, the numbers of EV-D68 infection and EV-D68-associated AFM cases rise biennially and it cannot be excluded that these will continue to rise, resulting in large outbreaks in the future. For a better understanding of the systemic pathogenesis of EV-D68-associated diseases, it is essential to continue the development of in vitro, ex vivo and in vivo models.
We have here discussed the different stages in the pathogenesis of EV-D68 infections in humans: respiratory infection, systemic dissemination and CNS invasion. However, how these different stages are related to the severity and spectrum of clinical disease is not yet fully understood. For example, it is currently unknown if systemic dissemination directly leads to CNS invasion, or if systemic dissemination occurs even during mild or subclinical disease. In addition, it is still unclear if EV-D68-associated paralysis is always the result of CNS invasion and infection of motor neurons, or whether EV-D68 invasion into the muscle tissues could directly result in paralysis. It is therefore important to get more insight into the systemic pathogenesis of EV-D68, including the cell and tissues tropism and the role of viral and host factors. This information will be essential to understand disease progression and how this can be detected early after disease onset. Furthermore, these studies will enable scientist to develop better and targeted intervention strategies for the wide spectrum of EV-D68 associated diseases.
Acknowledgments
This work was funded by a fellowship to DvR from the Netherlands Organization for Scientific Research (VIDI contract 91718308) and a EUR fellowship. SSNA received the Royal Thai Government Scholarship supported by the Ministry of Science and Technology of Thailand to perform her doctoral study. The funders play no role in the preparation and the completion of this review. The authors declare no conflict of interest.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Waghmare A, Pergam SA, Jerome KR, et al. Clinical disease due to enterovirus D68 in adult hematologic malignancy patients and hematopoietic cell transplant recipients. Blood. 2015;125(11):1724–1729.
- Giombini E, Rueca M, Barberi W, et al. Enterovirus D68-Associated Acute Flaccid Myelitis in Immunocompromised Woman, Italy. Emerg Infect Dis. 2017;23(10):1690–1693.
- Poelman R, Schuffenecker I, Van Leer-Buter C, et al. European surveillance for enterovirus D68 during the emerging North-American outbreak in 2014. J Clin Virol. 2015;71:1–9.
- Messacar K, Abzug MJ, Dominguez SR. The Emergence of Enterovirus-D68. Microbiol Spectr. 2016;4(3):3.
- Ayscue P, Van Haren K, Sheriff H, et al. Acute flaccid paralysis with anterior myelitis - California. MMWR Morb Mortal Wkly Rep. 2012 [June -Jun 2014];63(40):903–906.
- Esposito S, Chidini G, Cinnante C, et al. Acute flaccid myelitis associated with enterovirus-D68 infection in an otherwise healthy child. Virol J. 2017;14(1):4.
- Kreuter JD, Barnes A, McCarthy JE, et al. A fatal central nervous system enterovirus 68 infection. Arch Pathol Lab Med. 2011;135(6):793–796.
- Paul JR. Poliomyelitis attack rates in American troops, 1940-1948. Am J Hyg. 1949;50(1):57–62.
- Garon JR, Cochi SL, Orenstein WA. The Challenge of Global Poliomyelitis Eradication. Infect Dis Clin North Am. 2015;29(4):651–665.
- Messacar K, Schreiner TL, Maloney JA, et al. A cluster of acute flaccid paralysis and cranial nerve dysfunction temporally associated with an outbreak of enterovirus D68 in children in Colorado, USA. Lancet. 2015;385(9978):1662–1671.
- Top FH. Incidence of cranial nerve paralysis in poliomyelitis in relation to presence or absence of tonsils. II. In a largely rural area. Pediatrics. 1958;21(1):106–111.
- Baggen J, Thibaut HJ, Strating J, et al. The life cycle of non-polio enteroviruses and how to target it. Nat Rev Microbiol. 2018;16(6):368–381.
- Bal A, Sabatier M, Wirth T, et al. Emergence of enterovirus D68 clade D1, France, August to November 2018. Euro Surveill. 2019;24(3):3.
- Pellegrinelli L, Giardina F, Lunghi G, et al. Emergence of divergent enterovirus (EV) D68 sub-clade D1 strains, northern Italy, September to October 2018. Euro Surveill. 2019;24(7):7.
- Yip CCY, Lo JYC, Sridhar S, et al. First Report of a Fatal Case Associated with EV-D68 Infection in Hong Kong and Emergence of an Interclade Recombinant in China Revealed by Genome Analysis. Int J Mol Sci. 2017;18(5):5.
- Greninger AL, Naccache SN, Messacar K, et al. A novel outbreak enterovirus D68 strain associated with acute flaccid myelitis cases in the USA (2012-14): a retrospective cohort study. Lancet Infect Dis. 2015;15(6):671–682.
- Knoester M, Scholvinck EH, Poelman R, et al. Upsurge of Enterovirus D68, the Netherlands, 2016. Emerg Infect Dis. 2017;23(1):140–143.
- Hixon AM, Yu G, Leser JS, et al. A mouse model of paralytic myelitis caused by enterovirus D68. PLoS Pathog. 2017;13(2):e1006199.
- Rosenfeld AB, Warren AL, Racaniello VR. Neurotropism of Enterovirus D68 Isolates Is Independent of Sialic Acid and Is Not a Recently Acquired Phenotype. MBio. 2019;10(5):5.
- Sooksawasdi NAS, Meijer A, Bauer L, et al. Enhanced Enterovirus D68 Replication in Neuroblastoma Cells Is Associated with a Cell Culture-Adaptive Amino Acid Substitution in VP1. mSphere. 2020;5:6.
- Baggen J, HJ T, Staring J, et al. Enterovirus D68 receptor requirements unveiled by haploid genetics. Proc Natl Acad Sci U S A. 2016;113(5):1399–1404.
- Liu Y, Sheng J, Baggen J, et al. Sialic acid-dependent cell entry of human enterovirus D68. Nat Commun. 2015;6(1):8865.
- de Graaf M, Fouchier RA. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO J. 2014;33(8):823–841.
- Nicholls JM, Bourne AJ, Chen H, et al. Sialic acid receptor detection in the human respiratory tract: evidence for widespread distribution of potential binding sites for human and avian influenza viruses. Respir Res. 2007;8(1):73.
- Sauer AK, Liang CH, Stech J, et al. Characterization of the sialic acid binding activity of influenza A viruses using soluble variants of the H7 and H9 hemagglutinins. PLoS One. 2014;9(2):e89529.
- Brown DM, Hixon AM, Oldfield LM, et al. Contemporary Circulating Enterovirus D68 Strains Have Acquired the Capacity for Viral Entry and Replication in Human Neuronal Cells. mBio. 2018;9(5):5.
- Baggen J, Liu Y, Lyoo H, et al. Bypassing pan-enterovirus host factor PLA2G16. Nat Commun. 2019;10(1):3171.
- Tseligka ED, Sobo K, Stoppini L, et al. A VP1 mutation acquired during an enterovirus 71 disseminated infection confers heparan sulfate binding ability and modulates ex vivo tropism. PLoS Pathog. 2018;14(8):e1007190.
- Jenniskens GJ, Oosterhof A, Brandwijk R, et al. Heparan sulfate heterogeneity in skeletal muscle basal lamina: demonstration by phage display-derived antibodies. J Neurosci. 2000;20(11):4099–4111.
- Farhy Tselnicker I, Boisvert MM, Allen NJ. The role of neuronal versus astrocyte-derived heparan sulfate proteoglycans in brain development and injury. Biochem Soc Trans. 2014;42(5):1263–1269.
- Wei W, Guo H, Chang J, et al. ICAM-5/Telencephalin Is a Functional Entry Receptor for Enterovirus D68. Cell Host Microbe. 2016;20(5):631–641.
- Hixon AM, Clarke P, Tyler KL. Contemporary Circulating Enterovirus D68 Strains Infect and Undergo Retrograde Axonal Transport in Spinal Motor Neurons Independent of Sialic Acid. J Virol. 2019;93(16):16.
- Gahmberg CG, Tian L, Ning L, et al. ICAM-5–a novel two-facetted adhesion molecule in the mammalian brain. Immunol Lett. 2008;117(2):131–135.
- Itagaki T, Aoki Y, Matoba Y, et al. Clinical characteristics of children infected with enterovirus D68 in an outpatient clinic and the association with bronchial asthma. Infect Dis (Lond). 2018;50(4):303–312.
- Schuster JE, Selvarangan R, Hassan F, et al. Clinical Course of Enterovirus D68 in Hospitalized Children. Pediatr Infect Dis J. 2017;36(3):290–295.
- Schuster JE, Miller JO, Selvarangan R, et al. Severe enterovirus 68 respiratory illness in children requiring intensive care management. J Clin Virol. 2015;70:77–82.
- Korematsu S, Nagashima K, Sato Y, et al. “Spike” in acute asthma exacerbations during enterovirus D68 epidemic in Japan: a nation-wide survey. Allergol Int. 2018;67(1):55–60.
- Foster CB, Coelho R, Brown PM, et al. A comparison of hospitalized children with enterovirus D68 to those with rhinovirus. Pediatr Pulmonol. 2017;52(6):827–832.
- Kusabe Y, Takeshima A, Seino A, et al. [An Adult Case of Enterovirus D68 Encephalomyelitis Presenting as Bilateral Facial Nerve Palsy and Dysphagia]. Brain Nerve. 2017;69(8):957–961.
- Ward NS, Hughes BL, Mermel LA. Enterovirus D68 Infection in an Adult. Am J Crit Care. 2016;25(2):178–180.
- Matsumoto M, Awano H, Ogi M, et al. A pediatric patient with interstitial pneumonia due to enterovirus D68. J Infect Chemother. 2016;22(10):712–715.
- Essaidi-Laziosi M, Brito F, Benaoudia S, et al. Propagation of respiratory viruses in human airway epithelia reveals persistent virus-specific signatures. J Allergy Clin Immunol. 2018;141(6):2074–2084.
- Patel MC, Wang W, Pletneva LM, et al. Enterovirus D-68 Infection, Prophylaxis, and Vaccination in a Novel Permissive Animal Model, the Cotton Rat (Sigmodon hispidus). PLoS One. 2016;11(11):e0166336.
- Zheng HW, Sun M, Guo L, et al. Nasal Infection of Enterovirus D68 Leading to Lower Respiratory Tract Pathogenesis in Ferrets (Mustela putorius furo). Viruses. 2017;9(5):5.
- WJ E, BL H, CJ P, et al. Development of a respiratory disease model for enterovirus D68 in 4-week-old mice for evaluation of antiviral therapies. Antiviral Res. 2019;162:61–70.
- Rajput C, Han M, Bentley JK, et al. Enterovirus D68 infection induces IL-17-dependent neutrophilic airway inflammation and hyperresponsiveness. JCI Insight. 2018;3(16):16.
- Xiang Z, Liu L, Lei X, et al. 3C Protease of Enterovirus D68 Inhibits Cellular Defense Mediated by Interferon Regulatory Factor 7. J Virol. 2016;90(3):1613–1621.
- Rui Y, Su J, Wang H, et al. Disruption of MDA5-Mediated Innate Immune Responses by the 3C Proteins of Coxsackievirus A16, Coxsackievirus A6, and Enterovirus D68. J Virol. 2017;91(13):13.
- Kang J, Pang Z, Zhou Z, et al. Enterovirus D68 Protease 2A pro Targets TRAF3 To Subvert Host Innate Immune Responses. J Virol. 2021;95(3):3.
- Chong PF, Kira R, Mori H, et al. Clinical Features of Acute Flaccid Myelitis Temporally Associated With an Enterovirus D68 Outbreak: results of a Nationwide Survey of Acute Flaccid Paralysis in Japan, August-December 2015. Clin Infect Dis. 2018;66(5):653–664.
- Imamura T, Suzuki A, Lupisan S, et al. Detection of enterovirus 68 in serum from pediatric patients with pneumonia and their clinical outcomes. Influenza Other Respir Viruses. 2014;8(1):21–24.
- Hurst BL, Evans WJ, Smee DF, et al. Evaluation of antiviral therapies in respiratory and neurological disease models of Enterovirus D68 infection in mice. Virology. 2019;526:146–154.
- Zhang C, Zhang X, Dai W, et al. A Mouse Model of Enterovirus D68 Infection for Assessment of the Efficacy of Inactivated Vaccine. Viruses. 2018;10(2):2.
- Zheng H, Wang J, Li B, et al. A Novel Neutralizing Antibody Specific to the DE Loop of VP1 Can Inhibit EV-D68 Infection in Mice. J Immunol. 2018;201(9):2557–2569.
- Shen L, Chen CY, Huang D, et al. Pathogenic Events in a Nonhuman Primate Model of Oral Poliovirus Infection Leading to Paralytic Poliomyelitis. J Virol. 2017;91(14):14.
- Smura T, Ylipaasto P, Klemola P, et al. Cellular tropism of human enterovirus D species serotypes EV-94, EV-70, and EV-68 in vitro: implications for pathogenesis. J Med Virol. 2010;82(11):1940–1949.
- Chan MC, Chan RW, Yu WC, et al. Influenza H5N1 virus infection of polarized human alveolar epithelial cells and lung microvascular endothelial cells. Respir Res. 2009;10(1):102.
- Corona AK, Saulsbery HM, Corona Velazquez AF, et al. Enteroviruses Remodel Autophagic Trafficking through Regulation of Host SNARE Proteins to Promote Virus Replication and Cell Exit. Cell Rep. 2018;22(12):3304–3314.
- Liang CC, Sun MJ, Lei HY, et al. Human endothelial cell activation and apoptosis induced by enterovirus 71 infection. J Med Virol. 2004;74(4):597–603.
- Bozym RA, Morosky SA, Kim KS, et al. Release of intracellular calcium stores facilitates coxsackievirus entry into polarized endothelial cells. PLoS Pathog. 2010;6(10):e1001135.
- Conaldi PG, Serra C, Mossa A, et al. Persistent infection of human vascular endothelial cells by group B coxsackieviruses. J Infect Dis. 1997;175(3):693–696.
- Couderc T, Barzu T, Horaud F, et al. Poliovirus permissivity and specific receptor expression on human endothelial cells. Virology. 1990;174(1):95–102.
- Coyne CB, Kim KS, Bergelson JM. Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. Embo J. 2007;26(17):4016–4028.
- Saijets S, Ylipaasto P, Vaarala O, et al. Enterovirus infection and activation of human umbilical vein endothelial cells. J Med Virol. 2003;70(3):430–439.
- Wang W, Sun J, Wang N, et al. Enterovirus A71 capsid protein VP1 increases blood-brain barrier permeability and virus receptor vimentin on the brain endothelial cells. J Neurovirol. 2020;26(1):84–94.
- MM Z, Favaro E, PG C, et al. Persistent infection of human microvascular endothelial cells by coxsackie B viruses induces increased expression of adhesion molecules. J Immunol. 2003;171(1):438–446.
- Pham NTK, Thongprachum A, Baba T, et al. A 3-Month-Old Child with Acute Gastroenteritis with Enterovirus D68 Detected from Stool Specimen. Clin Lab. 2017;63(7):1269–1272.
- Van Haren K, Ayscue P, Waubant E, et al. Acute Flaccid Myelitis of Unknown Etiology in California, 2012-2015. Jama. 2015;314(24):2663–2671.
- Fall A, Ndiaye N, Jallow MM, et al. Enterovirus D68 Subclade B3 Circulation in Senegal, 2016: detection from Influenza-like Illness and Acute Flaccid Paralysis Surveillance. Sci Rep. 2019;9(1):13881.
- Williams CJ, Thomas RH, Pickersgill TP, et al. Cluster of atypical adult Guillain-Barre syndrome temporally associated with neurological illness due to EV-D68 in children, South Wales, United Kingdom, October 2015 to January 2016. Euro Surveill. 2016;21(4):4.
- Guo H, Li Y, Liu G, et al. A second open reading frame in human enterovirus determines viral replication in intestinal epithelial cells. Nat Commun. 2019;10(1):4066.
- Antona D, Kossorotoff M, Schuffenecker I, et al. Severe paediatric conditions linked with EV-A71 and EV-D68, France, May to October. Euro Surveill. 2016;21(46):46. 2016.
- Chang TH, Yang TI, Hsu WY, et al. Case report: painful exanthems caused by enterovirus D68 in an adolescent. Medicine (Baltimore). 2019;98(33):e16493.
- Midgley SE, Benschop K, Dyrdak R, et al. Co-circulation of multiple enterovirus D68 subclades, including a novel B3 cluster, across Europe in a season of expected low prevalence, 2019/20. Euro Surveill. 2020;25(2):2.
- Barnadas C, Midgley SE, Skov MN, et al. An enhanced Enterovirus surveillance system allows identification and characterization of rare and emerging respiratory enteroviruses in Denmark, 2015-16. J Clin Virol. 2017;93:40–44.
- Morrey JD, Wang H, Hurst BL, et al. Causation of Acute Flaccid Paralysis by Myelitis and Myositis in Enterovirus-D68 Infected Mice Deficient in Interferon alphabeta/gamma Receptor Deficient Mice. Viruses. 2018;10(1):1.
- Sejvar JJ, Lopez AS, Cortese MM, et al. Acute Flaccid Myelitis in the United States, August-December 2014: results of Nationwide Surveillance. Clin Infect Dis. 2016;63(6):737–745.
- Messacar K, Schreiner TL, Van Haren K, et al. Acute flaccid myelitis: a clinical review of US cases 2012-2015. Ann Neurol. 2016;80(3):326–338.
- Nelson GR, Bonkowsky JL, Doll E, et al. Recognition and Management of Acute Flaccid Myelitis in Children. Pediatr Neurol. 2016;55:17–21.
- Pfeiffer HC, Bragstad K, Skram MK, et al. Two cases of acute severe flaccid myelitis associated with enterovirus D68 infection in children, Norway, autumn 2014. Euro Surveill. 2015;20(10):21062.
- Maloney JA, Mirsky DM, Messacar K, et al. MRI findings in children with acute flaccid paralysis and cranial nerve dysfunction occurring during the 2014 enterovirus D68 outbreak. AJNR Am J Neuroradiol. 2015;36(2):245–250.
- Yogo N, Imamura T, Muto Y, et al. Cardiopulmonary failure as a result of brainstem encephalitis caused by enterovirus D68. BMJ Case Rep. 2019;12(11):11.
- Esposito S, Lunghi G, Zampiero A, et al. Enterovirus-D68 in the Cerebrospinal Fluid of Two Children with Aseptic Meningitis. Pediatr Infect Dis J. 2016;35(5):589–591.
- Lang M, Mirand A, Savy N, et al. Acute flaccid paralysis following enterovirus D68 associated pneumonia, France. Euro Surveill. 2014;19(44):44. 2014.
- Wali RK, Lee AH, Kam JC, et al. Acute Neurological Illness in a Kidney Transplant Recipient Following Infection With Enterovirus-D68: an Emerging Infection? Am J Transplant. 2015;15(12):3224–3228.
- Ruggieri V, Paz MI, Peretti MG, et al. Enterovirus D68 infection in a cluster of children with acute flaccid myelitis, Buenos Aires, Argentina, 2016. Eur J Paediatr Neurol. 2017;21(6):884–890.
- Sun S, Bian L, Gao F, et al. A neonatal mouse model of Enterovirus D68 infection induces both interstitial pneumonia and acute flaccid myelitis. Antiviral Res. 2019;161:108–115.
- Ohka S, Nihei C, Yamazaki M, et al. Poliovirus trafficking toward central nervous system via human poliovirus receptor-dependent and -independent pathway. Front Microbiol. 2012;3:147.
- Dyrdak R, Grabbe M, Hammas B, et al. Outbreak of enterovirus D68 of the new B3 lineage in Stockholm, Sweden, August to September. Euro Surveill. 2016;21(46):46. 2016.
- Piralla A, Principi N, Ruggiero L, et al. Enterovirus-D68 (EV-D68) in pediatric patients with respiratory infection: the circulation of a new B3 clade in Italy. J Clin Virol. 2018;99-100:91–96.
- Zhang Y, Cao J, Zhang S, et al. Genetic changes found in a distinct clade of Enterovirus D68 associated with paralysis during the 2014 outbreak. Virus Evol. 2016;2(1):vew015.
- Du J, Zheng B, Zheng W, et al. Analysis of Enterovirus 68 Strains from the 2014 North American Outbreak Reveals a New Clade, Indicating Viral Evolution. PLoS One. 2015;10(12):e0144208.
- Kim M, Yu JE, Lee JH, et al. Comparative analyses of influenza virus receptor distribution in the human and mouse brains. J Chem Neuroanat. 2013;52:49–57.
- Sakabe S, Iwatsuki-Horimoto K, Takano R, et al. Cytokine production by primary human macrophages infected with highly pathogenic H5N1 or pandemic H1N1 2009 influenza viruses. J Gen Virol. 2011;92(Pt 6):1428–1434.
- Videira PA, Amado IF, Crespo HJ, et al. Surface alpha 2-3- and alpha 2-6-sialylation of human monocytes and derived dendritic cells and its influence on endocytosis. Glycoconj J. 2008;25(3):259–268.
- Eash S, Tavares R, Stopa EG, et al. Differential distribution of the JC virus receptor-type sialic acid in normal human tissues. Am J Pathol. 2004;164(2):419–428.
- Yang H. Structure, Expression, and Function of ICAM-5. Comp Funct Genomics. 2012;2012:368938.
- Fadnes B, Husebekk A, Svineng G, et al. The proteoglycan repertoire of lymphoid cells. Glycoconj J. 2012;29(7):513–523.
- Wegrowski Y, Milard AL, Kotlarz G, et al. Cell surface proteoglycan expression during maturation of human monocytes-derived dendritic cells and macrophages. Clin Exp Immunol. 2006;144(3):485–493.
- Cabrerizo M, Garcia-Iniguez JP, Munell F, et al. First Cases of Severe Flaccid Paralysis Associated With Enterovirus D68 Infection in Spain, 2015-2016. Pediatr Infect Dis J. 2017;36(12):1214–1216.
- Hu YL, Huang LM, Lu CY, et al. Manifestations of enterovirus D68 and high seroconversion among children attending a kindergarten. J Microbiol Immunol Infect. 2019;52(6):858–864.
- ElBadry M, Lednicky J, Cella E, et al. Isolation of an Enterovirus D68 from Blood from a Child with Pneumonia in Rural Haiti: close Phylogenetic Linkage with New York Strain. Pediatr Infect Dis J. 2016;35(9):1048–1050.