
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The human pathogen Streptococcus pyogenes (Group A Streptococcus, GAS) is responsible for invasive disease characterized by inflammation and tissue destruction. Inflammatory symptoms of invasive disease may be attributed to the neutrophil response during the early stages of infection. Here, the human neutrophil response to GAS was characterized in vitro using emm98.1 type covS mutant GAS strain NS88.2 (isolated from invasive infection) and avirulent variant NS88.2rep. NS88.2 was shown to resist phagocytic killing and proliferate in the presence of human neutrophils, where neutrophil antimicrobial defence through the production of reactive oxygen species was reduced compared with NS88.2rep. In the presence of NS88.2, neutrophil death was delayed compared with NS88.2rep. Infection with either GAS strain induced expression of inflammatory caspases-1 and -4 in neutrophils, with increased detection of activated inflammatory caspases in response to NS88.2rep compared with NS88.2. NS88.2 infection caused differential expression of cell-surface CD66b, CD16, and CD31, when compared to NS88.2rep. We conclude that the neutrophil response to NS88.2 promotes inflammation and may be a contributing factor to the severity of invasive GAS infections.
Abbreviations
CovRS, control of virulence regulatory system; GAS, Group A Streptococcus; PMN, polymorphonuclear leukocyte.
Introduction
The human pathogen Streptococcus pyogenes (Group A Streptococcus, GAS) can cause invasive diseases with severe complications including necrotizing fasciitis, toxic shock, and sepsis [Citation1]. These life-threatening diseases progress swiftly and are characterized by uncontrollable inflammation and tissue degradation [Citation2–7]. Global surveillance studies in 2005 indicated that 163,000 deaths annually could be attributed to invasive GAS infection [Citation8]. Mortality of patients suffering toxic shock is between 23% and 81% in the first 7 days of infection [Citation1]. In high-income settings, the prevalence of serotype emm1 (M1) GAS in invasive disease is high [Citation4,Citation5,Citation9,Citation10]. In particular, the emm1 GAS clone M1T1 has globally disseminated, causing disease through immune hyperstimulation and resistance to host neutrophil killing [Citation11]. The M1T1 clone is also prone to genetic mutation in the control of virulence regulator system sensor (covS), leading to hypervirulence in animal models of GAS infection [Citation12]. A large body of research has focused on emm1 GAS, but in demographics with increased risk and burden of infection, GAS serotype distribution is far more diverse with no distinct dominant strains [Citation13–15]. Other GAS emm types harboring covS mutations (such as emm98.1 GAS strain NS88.2) display similar resistance to neutrophil killing and also have a high propensity to cause invasive disease in mouse models of GAS infection [Citation16,Citation17]. However, in comparison to emm1, non-emm1 pathogenic mechanisms and host immune responses are less well-defined.
Neutrophils (polymorphonuclear leukocytes, PMNs) play an essential part in host defence against bacterial pathogens. Bacterial resistance to or evasion of neutrophils can promote disease [Citation18]. Neutrophils are abundant at sites of invasive GAS infection, where they contribute to pathogen removal through a sequence of phagocytosis, bactericidal activity mediated by cytotoxic contents, then cell-death [Citation19–21]. During this process, neutrophils down-regulate their inflammatory response and signal for their clearance by macrophages through the process of efferocytosis [Citation22,Citation23]. The safe removal of neutrophils prevents unwanted leakage of cytotoxic molecules, ensuring no damage to the surrounding tissue [Citation24]. Alternative pro-inflammatory neutrophil pathways which involve cell lysis such as pyroptosis and necroptosis, may also be induced in response to bacteria [Citation25]. Unregulated pro-inflammatory pathways, however, pose a threat to the host through the release of cytotoxic mediators to initiate a cascade of inflammation and tissue destruction [Citation26,Citation27].
Infection control by neutrophils is thought to be a homoeostatic relationship between anti-inflammatory and pro-inflammatory pathways, where disruption of neutrophil-mediated immunity may be a contributing factor to the development of invasive disease [Citation24]. Neutrophil disruption and inflammatory death have been reported in response to GAS strains isolated from invasive infections (emm1 and emm98.1) [Citation17,Citation28,Citation29]. Characterization of the neutrophil response to emm1 GAS suggests that inflammatory neutrophil death and disruption to neutrophil phagocytosis may reduce bacterial clearance and promote an inflammatory neutrophil phenotype [Citation29]. It is unclear if this response is emm1-specific or characteristic of other GAS serotypes isolated from invasive infection. Previous work investigating emm98.1 (NS88.2) GAS infection of neutrophils identified a lytic form of cell death [Citation17], but the neutrophil phenotype was largely uncharacterised. The current study uses an in vitro human neutrophil model of infection to report the effect of emm98.1 GAS upon neutrophil death, signaling, and associated inflammatory profile. This study builds on the limited body of work investigating the host response to infection due to covS mutation using emm98.1 GAS and in a corresponding isogenic strain in which a covS mutation has been repaired [Citation16,Citation17]. We hypothesize that NS88.2 disrupts processes that promote the resolution of infection, decreasing neutrophil antibacterial function. Investigation of non-emm1 GAS is essential to determine if there is a common neutrophil phenotype during invasive infection, as this may have implications for the application of new therapeutics.
Materials and methods
Ethics statement
The following procedures were reviewed, authorized and approved by the University of Wollongong Human Ethics Committee (Protocol HE08/250). Healthy volunteers (female and male of differing age) provided informed consent prior to the donation of limited venous blood for experiments involving human neutrophils.
Bacterial strains and culture
Escherichia coli MC1061 were grown in Luria – Bertani Broth (LB) at 37°C with constant shaking. S. pyogenes clinical bacteremia isolate NS88.2 (emm98.1, containing a non-functional mutation to the control of virulence regulator system (covRS) sensor covS) and isogenic mutant with functional covRS NS88.2covSrep (NS88.2rep) have been described previously [Citation16]. GAS were cultured from storage at 37°C on horse-blood agar (Oxoid, PP2001). For experimental enumeration of colony forming units (CFU)/mL, GAS were but cultured on 1% yeast supplemented (w/v) Todd-Hewitt broth (THY, Bacto Laboratories 249,240) agar. For in vitro infection, GAS were cultured overnight at 37°C in THY, then sub-inoculated (1:10) into fresh THY and grown to mid-logarithmic phase. When required, antibiotics were added to THY (100 μg/ml ampicillin and 200 μg/ml kanamycin). GAS were washed twice with phosphate buffered saline (PBS) before resuspension at the desired multiplicity of infection (MOI). GAS green fluorescent protein (GFP) expression was achieved by transforming GAS with pLZ12Km2-P23R-TA:GFP [Citation29] using electroporation [Citation30]. Expression of GFP by GAS transformants was confirmed with flow cytometry.
Isolation of human neutrophils
Venous human blood was collected in 10 mL lithium heparin-coated Vacutainer (BD 367,526) tubes, allowed to sit and cool to room temperature (RT), then carefully layered over an equal volume of RT Polymorphprep (Axis Shield 1,114,683) and centrifuged as per the manufacturer’s instructions. Polymorphprep is well established to demonstrate high neutrophil yield with low contamination with other leukocytes [Citation31]. Neutrophils were isolated and any remaining erythrocytes hypotonically lysed. Purified neutrophils were resuspended in Hank’s Balanced Salt solution (without Ca2+ or Mg2+, Corning Inc., 55–022-PBR). For in vitro infection, neutrophils were resuspended in complete medium, Roswell-Park Memorial Institute (RPMI) medium 1640 (Gibco 11,875,101)) containing 2% heat-inactivated autologous plasma (v/v) and 2 mM L-glutamine (Gibco 25,030,149) at the desired concentration, unless stated otherwise. Neutrophil viability was assessed via staining with Trypan Blue (Sigma-Aldrich, T8154-100 ML) staining was used to confirm neutrophil viability, while purity was determined using a BD LSR Fortessa X-20 flow cytometer via assessment of forward and side scatter profiles. Peridinin chlorophyll protein (PerCP)/Cy5.5-conjugated anti-CD66b antibody (clone G10F5, BioLegend 305,102) was routinely expressed on over 95% of purified cells. Neutrophils were kept at RT throughout processing to preserve viability.
Flow cytometry
Data collection was performed using a BD LSR Fortessa X-20 flow cytometer with excitation lasers: 405 nm; 488 nm; 561 nm; and 640 nm. Bandpass filters used were: Zombie Aqua Fixable Viability Dye (525/50); fluorescein isothiocynate (FITC)/GFP/FAM FLICA (525/50); PerCP-Cy5.5 (695/40) R-phycoerythrin (PE, 586/15); PE-Cy7 (780/60); and allophycocyanin (APC, 670/30). FlowJo software V10.6.1 (TreeStar Inc.) was used for the analysis of flow cytometry data.
In vitro infection of neutrophils with Group A streptococcus
Following purification, GAS and neutrophils were co-cultured at varying MOI (shown as GAS:neutrophil) and incubated at 37°C in 5% CO2.
Group A streptococcus phagocytosis
GAS expressing GFP were co-cultured with neutrophils (24-well plate at MOI 10:1) in complete medium. Cells were washed with 10% heat-inactivated fetal bovine serum (FBS, Bovogen Biologicals, SFBS-AU) (v/v) in PBS then analyzed for phagocytosis using flow cytometry.
Group A streptococcus internalisation by neutrophils (gentamicin protection assay)
GAS were co-cultured with neutrophils (96-well plate at MOI 50:1) for 30 min at 37°C in 5% CO2. Cells were washed thrice with complete medium, hypotonically lysed with water and serially diluted before plating to enumerate bacterial concentrations for associated GAS or washed once with complete medium, resuspended in complete medium containing 200 µg/mL gentamicin and incubated for a further 60 min at 37°C in 5% CO2. Neutrophils were then washed thrice with complete medium then hypotonically lysed and serially diluted before plating to enumerate bacterial concentrations. Internalization was determined as GAS concentration after antibiotic treatment over total GAS concentration associated to neutrophils, using the formula:
Neutrophil production of reactive oxygen species
The production of reactive oxygen species (ROS) by neutrophils was assessed as described [Citation28]. In short, neutrophils were pre-incubated in complete medium containing 25 μM 2,’7’-dichlorodihyrdofluorescein diacetate (DCF, Molecular Probes) for 45 min at RT prior to co-culture with GAS (96-well plate at MOI 10:1). Rate of ROS production was determined by measuring fluorescence (ex485 nm em520 nm) between 30 and 60 min at 37°C using a POLARstar Omega plate reader (BMG Labtech, Ortenberg, Germany).
Group A streptococcus survival and proliferation
GAS were co-cultured with neutrophils (96-well plate at MOI 1:10) or lysed via three freeze-thaw (using liquid N2) cycles prior to co-culture. Where required, neutrophil phagocytosis was inhibited by preincubating neutrophils with 10 μm cytochalasin D (Cayman Chemical 11,330) for 30 min at 37°C in complete medium prior to co-culture. Following hypotonic lysis of neutrophils, lysates were serially diluted and plated on THY agar. GAS survival was determined by comparing the CFU/mL in the presence of absence of neutrophils, using the formula:
Neutrophil viability
GAS were co-cultured with neutrophils (24-well plate at MOI 10:1) and neutrophil viability was assessed. Cells were washed with PBS then stained with Zombie Aqua membrane viability dye (BioLegend 423,102) for 15 min at RT. Cells were washed in PBS, then in 10% FBS (v/v) in PBS and incubated with annexin-V-FITC (BioLegend 640,945) for 15 min at RT then analyzed immediately using a flow cytometer. Neutrophil viability was reported as a percentage of total neutrophils.
Immunoblotting and antibodies
Following GAS co-culture (6-well plate at MOI 10:1), neutrophils were lysed using RIPA buffer (150 mM NaCl, 5 mM EDTA, 50 mM Tris, 1.0% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate (all v/v), 2 × cOmplete protease inhibitor cocktail (Roche 4,693,132,001), 1 mM phenylmethylsulphonyl fluoride, 5 mM sodium pyrophosphate, 5 mM sodium molybdate and 5 mM β-glycerophosphate) as described [Citation29]. Twenty μg of lysate was separated by electrophoresis (4–20% TGX Stain-Free protein gels, Bio-Rad 4,568,095) and total protein was determined (Bio-Rad ChemiDoc XR, Image Lab Software) before immunoblotting to 0.2 μm Immun-Blot PVDF (Bio-Rad 1,620,177). PVDF membranes were probed with caspase-1 polyclonal (1:1000, 2225s), caspase-3 polyclonal (1:1000, 9662), caspase-4 polyclonal (1:1000, 4450s) or caspase-8 monoclonal (1:1000, 4790s) primary antibodies (Cell Signalling Technology) and horse radish peroxidase-conjugated goat anti-rabbit IgG (1:5000, Invitrogen, 65–6120) secondary antibody. Clarity or Clarity Max Western ECL Blotting Substrates (Bio-Rad 1,705,060 or 1705062S) were used for detection and imaged with an Amersham AI600 (GE Healthcare). Area under the peak was quantified for each band and normalized over total lane protein using the ImageJ software (National Institutes of Health).
FAM-FLICA inflammatory caspase activation
Inflammatory caspase activation (caspase-1, -4, and -5) was assessed during GAS co-cultured with neutrophils (24-well plate at MOI 10:1) using the FAM-FLICA Caspase-1 assay kit (ImmunoChemistry Technologies, 98), as described [Citation29] and in line with the manufacturer’s instructions. Inflammatory caspase activation was measured using flow cytometry.
Cytometric cytokine bead assay
Cytokine release was measured in supernatant following GAS co-culture with neutrophils (96-well plate at MOI 10:1) over 360 min using the LEGENDplex™ Human Inflammation Panel bead-based immunoassay (BioLegend 740,809) and flow cytometry, in line with the manufacturer’s instructions. Additional experiments were performed with a custom panel for interleukin (IL)-1β, IL-8 and tumour necrosis factor (TNF)-α release only. Flow cytometry data were analyzed using the LEGENDplex™ software V8.0 (VigeneTech Inc.).
Cluster of differentiation expression
Cell surface CD expression was measured on neutrophils following GAS co-culture (24-well plate at MOI 10:1). The cells were washed with PBS then stained with Zombie Aqua Fixable Viability dye for 15 min at RT. Cells were washed in PBS, followed by 10% FBS (v/v) in PBS. Cells were incubated with fluorochrome-conjugated antibodies CD11b-FITC (Clone ICRF44, 301330), CD31-PE/Cy7 (Clone WM59, 303118), or CD66b-PerCP/Cy5.5 (Clone G10F5, 305108) (BioLegend) and CD16-FITC (Clone 3G8, BD 556,618) for 15 min at RT. Cells were washed with 10% FBS (v/v) in PBS before analysis with a flow cytometer.
Statistical analyses
Prism 6 (GraphPad Software Inc.) and IBM SPSS Statistics 25 (IBM corporation) were used for data analysis and data visualisation was performed in Prism 6. Data were compared using a one-way or two-way ANOVA where differences were elucidated using Tukey’s HSD or Holm-Šídák () test, as appropriate. In two instances a Student’s t-test was used (). Neutrophil annexin-V/Zombie () and CD expression () data were analyzed using a linear mixed model to determine the significant interaction between treatment and time, due to the nested nature of repeated measurements over time in the same donor. Post-hoc tests were performed for the linear mixed model by Multiple one-way ANOVA were performed as post-hoc tests for the linear mixed model at each time point, where differences were elucidated using Tukey’s HSD test. Post-hoc tests were only applied to significant interactions. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Figure 1. GAS persistence and proliferation occurs due to reduced phagocytosis and internalisation. (a) GFP GAS association with human neutrophils after 30 min incubation (n = 6 donors, Student’s t-test) via flow cytometry (Figure S1). (b) The internalisation of GAS by neutrophils determined by gentamicin protection assay (n = 5 donors, Student’s t-test). (c) Rate of ROS production by neutrophils between 30 and 60 min (n = 3 donors). GAS strains were incubated in the presence of (d) active human neutrophils (n = 8 donors) and (E) lysed neutrophils (n = 4 donors) over 180 min and percent survival determined. (F) GAS killing by human neutrophils at 30 min following pre-incubation with cytochalasin D to inhibit phagocytosis (n = 4 donors). (c-f) Holm-Šídák multiple comparison. Results are the pooled means ± SD of triplicate measurements. *p < 0.05, **p < 0.01 and ****p < 0.0001.
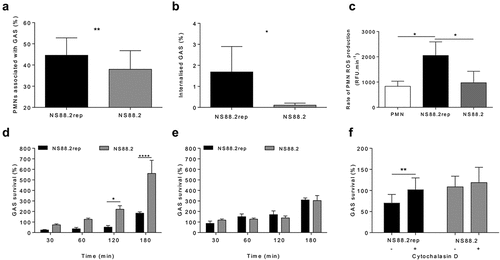
Figure 2. GAS infection induces neutrophil death. Human neutrophils (PMNs) were incubated with NS88.2, NS88.2rep or in the absence of GAS and stained with annexin V-FITC (AV) and Zombie Aqua viability dye (Z) before being analysed by flow cytometry. Neutrophils were gated as (a) singlets and then total (b) neutrophils (live and dead). Cells were analysed by fluorescence of annexin V-FITC and Zombie Aqua binding, where flow cytometry plots are representative and show (c) uninfected neutrophils, (d) NS88.2rep infected neutrophils and (e) NS88.2 infected neutrophils at 30 min. (f) Neutrophils were sampled over 180 min and the percentage of viable (AV−Z−) neutrophils are shown. Results are pooled means ± SD (n = 3 donors). Linear mixed model “p” values for treatment and time are stated. *p < 0.05, ***p < 0.001 and ****p < 0.0001, black denotes significance from control and grey denotes between NS88.2rep and NS88.2.
Results
Group A Streptococcus persistence occurs during a dampened neutrophil phagocytic response
Neutrophil-mediated phagocytosis and killing plays an important role in the removal of bacterial pathogens [Citation18]. Previous work has shown that the covS mutant NS88.2 is resistant to neutrophil-mediated killing when compared to the functional covS derivative NS88.2rep [Citation17]. In accordance with previous findings [Citation17], NS88.2 showed reduced association to human neutrophils compared to NS88.2rep following a 30 min infection (p < 0.01, and S1). Internalization of NS88.2 by neutrophils was also reduced compared to NS88.2rep (p < 0.05, ). Moreover, NS88.2rep induced significantly increased neutrophil ROS production compared to NS88.2, where a decreased rate of ROS production suggests a dampened antibacterial response ().
To expand our understanding of the neutrophil–GAS interaction, GAS survival in the presence of neutrophils was monitored over a 180 min time period. Increased survival of NS88.2 during neutrophil challenge was observed over this time period when compared to NS88.2rep (p < 0.05, ). Further, in the presence of viable human neutrophils NS88.2 showed increased proliferation compared to NS88.2rep (). No difference in proliferation between strains was detected when GAS was cultured with neutrophil lysates (). Finally, neutrophil pre-incubation with cytochalasin D, a phagocytosis inhibitor, resulted in increased NS88.2rep survival (p < 0.01), but not NS88.2 survival over 30 min (). Collectively, these data support previous work [Citation17] suggesting that increased survival of NS88.2 can be attributed in part, to reduced phagocytic function and ROS production.
Neutrophil death is delayed in response to NS88.2
Cell death in response to GAS infection has been described [Citation32,Citation33], however comparatively fewer studies have focused on neutrophils compared to other immune cell types [Citation17,Citation28,Citation29]. The exposure of phosphatidylserine (PS) on the cell-surface is a common hallmark of numerous cell-death pathways [Citation34]. To investigate GAS-induced neutrophil death, human neutrophils were incubated in the presence or absence of NS88.2rep or NS88.2, where annexin-V (AV) binding (measuring exposure of PS) and staining with Zombie Aqua viability dye (Z, measuring membrane integrity), was assessed flow cytometrically (). Neutrophils alone remained > 80% viable over 180 min (). In contrast, the incubation of neutrophils with either NS88.2rep or NS88.2 resulted in reduced neutrophil viability (p < 0.05), with significantly reduced viability following NS88.2rep infection compared to NS88.2 infection (p < 0.001, ). Results indicate that induction of neutrophil death is delayed in response to covS mutant NS88.2, compared to NS88.2rep, suggesting extended neutrophil viability during early infection.
Caspase-1 and caspase-4 are upregulated in neutrophils in response to emm98.1 GAS
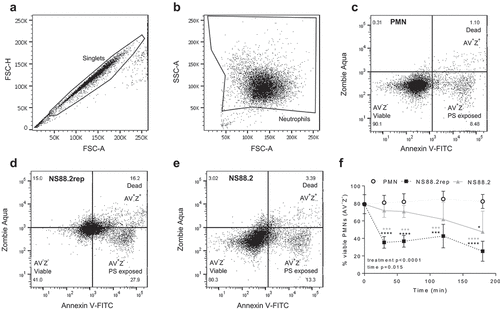
To further explore neutrophil-death occurring during GAS infection (), human neutrophil lysates were separated via protein electrophoresis then the abundance of active caspase-3, caspase-8, caspase-1 and caspase-4 was assessed by immunoblotting ( and S2–4). Caspase-3 and caspase-8 are critical in neutrophil apoptosis [Citation35], whereas caspase-1 and caspase-4 indicate inflammasome activation and can have downstream effects that induce pyroptosis [Citation36,Citation37]. Uninfected neutrophils displayed limited caspase-3 cleavage and decreasing amounts of caspase-8 over 180 min ( and S2A-C). Infection of neutrophils with NS88.2rep significantly increased caspase-3 p17 (p < 0.0001), and full-length caspase-8 (p < 0.01) concentrations compared to uninfected and NS88.2 infected neutrophils ( and S2b-c). Uninfected neutrophils displayed reduced amounts of caspase-1 and caspase-4 over 180 min ( and S2d-f). Infection of neutrophils with GAS increased pro-caspase-1 compared to uninfected neutrophils, however these differences were not statistically significant ( and S2d). Increased amounts of caspase-1 p46 were detected during infection with both GAS strains when compared to uninfected neutrophils (p < 0.0001). Notably, NS88.2rep infection invoked greater amounts of caspase-1 p46 compared to NS88.2 (p < 0.05, and S2e). Increased amounts of full-length caspase-4 were detected in neutrophils during infection with both GAS strains compared to uninfected cells (p < 0.001), and again, NS88.2rep invoked greater amounts of caspase-4 compared to NS88.2 (p < 0.01, and S2f). Thus, in addition to delayed death, neutrophils infected with NS88.2 show a reduction in the detectable levels of multiple caspases compared to NS88.2rep.
Figure 3. GAS infection increases inflammatory caspase abundance and activation in human neutrophils and induces IL-1β and TNF-α release. (a) Human neutrophil (PMN) lysates uninfected and (30, 60 and 180 min) during GAS infection immunoblotted for caspase-3, caspase-8, caspase-1, caspase-4 and GAPDH. Sample images from a single donor are displayed and represent triplicate experiments (Figures S3 and S4). SE=short exposure, LE=long exposure. Bands were quantified (ImageJ) and normalised against total protein (Figure S2). (b) Neutrophil inflammatory caspase activation during GAS infection using FLICA (FAM-YVAD-FMK) via flow cytometry (n = 3 donors). Singlets/PMNs (Figures S5A and B) and FAM-FLICA fluorescence (Figure S5C) was used for gating. Neutrophil cytokine release in the presence or absence of GAS was measured (0, 60, 180 and 360 min) using the LEGENDplex™ human inflammation cytometric bead assay. Neutrophils differentially released (c) IL-1β, (d) TNF-α and (e) IL-8 in response to GAS infection (duplicate measurements, n = 6 donors). Results are the pooled means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
To further explore caspase activation in response to GAS, inflammatory caspase activation was measured using a fluorogenic flow cytometric assay ( and S5). Uninfected neutrophils exhibited decreasing inflammatory caspase activation over time, however a significant interaction was only determined between treatments, not time (). NS88.2rep invoked increased inflammatory caspase activation at all time points when compared to uninfected or NS88.2 infected neutrophils (). In contrast, NS88.2 infection invoked increased inflammatory caspase activation at 180 min only (p < 0.01). Thus, neutrophil death following emm98.1 infection is accompanied by increased expression of caspase-1 and caspase-4, and inflammatory caspase activation, but this response is less evident in the presence of covS mutant strain NS88.2.
Neutrophils release IL-1β, TNF-α, and IL-8 during infection with emm 98.1 GAS
During invasive GAS infection, increased cytokine concentration negatively correlates to disease severity and patient outcomes [Citation38]. Specifically, IL-1β release can occur due to caspase-1 activation and is often associated with pyroptosis [Citation39]. Therefore, to investigate the contribution neutrophils play in cytokine production, human neutrophils were incubated with or without NS88.2rep or NS88.2 and supernatants analysed for cytokines (). Uninfected neutrophils released negligible amounts of IL-1β () and TNF-α (), however IL-8 release increased over 360 min (). In contrast, the incubation of neutrophils with GAS, invoked IL-1β release at 360 min (p < 0.0001), with significantly more IL-1β released in response to NS88.2rep compared to NS88.2 (p < 0.0001, ). Neutrophils also released TNF-α in response to both GAS strains at 180 and 360 min (p < 0.01), where again significantly more TNF-α was released in response to NS88.2rep compared to NS88.2 at 360 min (p < 0.0001, ). Increased IL-8 was detected at 60 min in response to NS88.2rep, when compared to NS88.2 infected or uninfected cells (p < 0.05, ). Collectively, these results indicate the release of inflammatory cytokines IL-1β and TNF-α in response to GAS infection, with an increased response seen during NS88.2rep infection.
Neutrophil cell-surface CD expression is altered in response to NS88.2
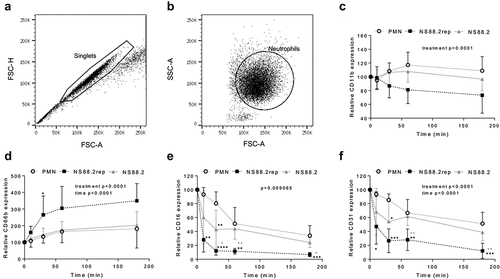
CD11b (Mac-1) and CD66b (CEACAM8) have important roles in innate immunity, responsible for neutrophil adhesion and activation, respectively [Citation40–42]. Opsonization of pathogens is facilitated by CD16 (FcγRIII), though it is downregulated during apoptosis [Citation43,Citation44]. Down-regulation of CD31 (PECAM-1), however, signals for the clearance of apoptotic neutrophils and has an anti-inflammatory influence [Citation45,Citation46]. Neutrophil 470 function was further explored following incubation with NS88.2, NS88.2 rep or in the absence of GAS by measuring the expression of cell-surface CD11b, CD66b, CD16 and CD31 (). In the absence of GAS, or following incubation with NS88.2, neutrophils exhibited slight increases in CD11b expression over time. In contrast, CD11b expression decreased during infection with NS88.2rep, though differences were not statistically significant upon post-hoc analysis (). Infection with NS88.2 rep significantly increased CD66b expression by neutrophils 30 min post-infection in comparison to both untreated and NS88.2 infected neutrophils, though a statistical difference was only determined between NS88.2rep infected and uninfected neutrophils (p < 0.05, ). Infection with NS88.2rep resulted in a significant decrease in CD16 expression over time compared to both neutrophils alone and those infected with NS88.2 at 30 (p < 0.05), 60 (p < 0.01), and 180 min (p < 0.05) post infection (). Similarly, to CD16, infection of neutrophils with NS88.2rep significantly reduced CD31 expression, at 60 and 180 min (p < 0.01) compared to NS88.2 (). Together, these data suggest that the neutrophil response to NS88.2 is dampened and similar to uninfected neutrophils, whereas NS88.2rep infection invokes significant changes to CD expression at the neutrophil cell-surface. Differences in cell-surface CD expression may be attributed to ineffective neutrophil activation and phagocytosis of NS88.2.
Figure 4. GAS infection influences neutrophil functionality. Human neutrophils (PMNs) were incubated in the absence or presence of GAS and cell-surface CD expression assessed over 180 min by flow cytometry. Neutrophils were sequentially gated as (a) singlets then (B) viable neutrophils, where plots (a-b) are representative and show NS88.2 infected neutrophils at 30 min. Neutrophil (c) CD11b-FITC (n = 4 donors), (d) CD66b-PerCP/Cy5.5 (n = 5 donors), (e) CD16-FITC (n = 4 donors) and (F) CD31-PE/Cy7 (n = 4 donors) was quantified for each donor as relative mean fluorescence intensity. Results are the pooled means ± SD. Linear mixed model “p” values represent interaction (treatment*time) or are stated. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001, with black denoting significance from control and grey between NS88.2rep and NS88.2.
Discussion
The response of neutrophils to pathogens contributes significantly to the outcomes of infection [Citation18]. Previous work has identified a lytic form of neutrophil death in response to emm98.1 GAS [Citation17]. The current study furthers the understanding of the human neutrophil response to GAS, characterizing neutrophil death, cell-surface signaling and inflammatory profile during the early stages of emm98.1 (NS88.2 and NS88.2rep) infection in vitro. Neutrophils can invoke an inflammatory phenotype in response to GAS infection, as evidenced by increased casapase-1 and caspase-4 expression and inflammatory caspase activation during cell death in vitro. Further, GAS harboring covS mutations have increased survival and proliferation in the presence of human neutrophils. We determine that in the GAS emm98.1 genetic background, covS mutation influences inflammatory caspases-1/-4/-5 activation and release of IL-1β by neutrophils. For NS88.2rep, expressing a functional covS, phagocytosis by neutrophils is followed by substantial ROS production and rapid induction of neutrophil death, indicated by cleaved caspase-3, caspase-1 and caspase-4. Additionally, reduced neutrophil viability following NS882.rep phagocytosis, coincides with the decreased expression of CD16 and CD31. In contrast, covS mutant NS88.2 instigates limited neutrophil activation and production of ROS, resulting in extended neutrophil survival and reduced GAS killing. CD16 and CD31 are retained on the neutrophil cell-surface during infection with NS88.2, which may hinder the removal of GAS and neutrophil clearance at sites of infection, delaying the resolution of innate-mediated inflammation.
The induction of lytic neutrophil death, as inferred by loss of cell membrane integrity, during emm1 and emm98.1 GAS infection has been reported previously [Citation17,Citation28]. The current study identifies the activation of inflammatory caspases in neutrophils during emm98.1 GAS infection, supporting a hypothesis that an inflammatory regulated pathway may be activated. Inflammatory caspase activation was more evident for NS88.2rep than the covS mutant NS88.2, which may be attributed to differences in observed rates of phagocytosis. Increased release of neutrophil activation molecule IL-8 [Citation47] seen during NS88.2rep infection may aid ROS production [Citation48] and further support differences in phagocytic killing. It is hypothesized that reduced ROS release increases the ability of NS88.2 to resist phagocytosis, which has been reported previously [Citation17], and in the current study, reduced phagocytic uptake also correlates with delayed neutrophil death. Similarly, the release of inflammatory cytokines was different between the two strains, where increased inflammatory caspase activation correlates with increased release of IL-1β. Reduced expression of caspase-3 and caspase-8 during NS88.2 infection was also observed and is indicative of reduced neutrophil apoptosis [Citation35]. A homoeostatic relationship between apoptotic and inflammatory pathways is thought to effectively facilitate the removal of bacterial pathogens and control inflammation [Citation24]. Disruption of this homeostatic balance, as demonstrated during NS88.2 infection, may therefore be a contributing factor to the development of invasive GAS disease.
Multiple GAS virulence factors confer resistance to neutrophil-mediated killing [Citation1], and it is well-established that innate immune responses can dictate infection outcomes [Citation18,Citation24]. In the current study, limited increases in CD66b expression on cells infected with NS88.2 indicate low neutrophil activation and production of ROS for oxidative burst [Citation42,Citation49]. Furthermore, neutrophils showed rapid loss of CD16 and CD31 during infection with NS88.2rep, whilst neutrophils infected with NS88.2 retained these markers over time. CD31 retention, as observed for neutrophils infected with NS88.2, can hinder the process of efferocytosis [Citation45], limiting neutrophil removal by macrophages [Citation46] and contributing to cellular crowding at the site of infection. Infection of neutrophils with gram-positive cocci Staphylococcus aureus has been shown to prevent macrophage efferocytosis [Citation50]. Neutrophil CD31 retention may have a similar effect during GAS infection, although this was not explored in the current study. Moreover, neutrophils infected with NS88.2 retained CD16, which functions as a receptor for immune complexes and opsonised bacteria, so this may indicate a reduction in phagocytic function and apoptosis. Collectively, these data show that the neutrophil response to covS mutant GAS is altered in a way that may favour a prolonged inflammatory response at sites of infection.
Reports for other pathogens, such as Pseudomonas aeruginosa and S. aureus describe alteration to neutrophil function and inflammatory death as contributing factors to disease development [Citation51–53]. In murine neutrophils, virulent GAS survive intracellularly, evading host defences and using the inflammatory response for a competitive advantage to disseminate and increasing virulence [Citation54,Citation55]. These and other studies support the hypothesis that neutrophil death may also contribute to the development of invasive GAS disease [Citation17,Citation28]. Both emm98.1 (this study) and emm1 [Citation29] GAS induce caspase-1 activation and cytokine release from neutrophils, while reduced neutrophil apoptosis occurs in response to strains that are highly virulent in vivo and harbour covS mutations [Citation17,Citation29]. A homoeostatic relationship between apoptotic and inflammatory neutrophil-death is believed to determine the control of bacterial infections [Citation18,Citation24]. Disruption to this relationship may be a common phenotype of severe invasive GAS infections.
In summary, the neutrophil response plays an important role in the removal of bacteria, where the function of neutrophils during the early stage of infection can greatly influence outcomes. Here, evidence is provided to show that, in line with what has been reported for emm1 GAS, neutrophil function is altered during infection with emm98.1 GAS strain NS88.2, isolated from invasive disease. Alterations to anti-inflammatory pathways inferred through cell-surface CD markers, may hinder the resolution of infection, whilst inflammatory neutrophil death and cytokine release promote further inflammation. The demonstration that diverse GAS serotypes are associated with caspase-1 activation in neutrophils supports the need to further explore inflammasome activation in response to GAS. Disruption to the equilibrium of apoptotic and inflammatory neutrophil death may be a common mechanism occurring during invasive infections. Additionally, targeted therapies may aim to not only control inflammatory neutrophil-death but also encourage the induction of anti-inflammatory pathways to limit severe pathologies that contribute to invasive GAS disease progression.
Author contributions
JW: Conceptualization, methodology, formal analysis, investigation, writing – original draft, writing – review and editing, visualization. RS: Conceptualization, methodology, writing – review and editing, visualization, supervision, funding acquisition. MSS: Conceptualization, methodology, resources, writing – review and editing, visualization, supervision, funding acquisition. All authors adhere to ICMJE authorship guidelines and have approved the submitted version for publication.
Supplemental Material
Download MS Word (8.1 MB)Acknowledgements
We thank all the generous participants for their sample donation and time. We thank the Molecular Horizons Fluorescence Analysis Facility at the University of Wollongong. This project was funded by the Illawarra Health and Medical Research Institute 2018 NHMRC Near Miss Scheme, awarded to MSS and RS. JW was a recipient of the Research Training Program Scholarship during this time.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The raw data supporting the conclusions of this article are not freely available due to limits of ethics approval, however project requests can be made by contacting the corresponding author for access and amendments on a case-by-case basis.
Supplemental data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2023.2264090.
Additional information
Funding
References
- Walker MJ, Barnett TC, Mcarthur JD, et al. Disease manifestations and pathogenic mechanisms of group a Streptococcus. Clin Microbiol Rev. 2014;27(2):264–12. doi: 10.1128/CMR.00101-13
- Davies HD, Mcgeer A, Schwartz B, et al. Invasive group a streptococcal infections in Ontario, Canada. N Engl J Med. 1996;335(8):547–554. doi: 10.1056/NEJM199608223350803
- Lamagni TL, Darenberg J, Luca-Harari B, et al. Epidemiology of severe Streptococcus pyogenes disease in Europe. J Clin Microbiol. 2008;46:2359–2367. doi: 10.1128/JCM.00422-08
- Nelson GE, Pondo T, Toews KA, et al. Epidemiology of invasive group a streptococcal infections in the United States, 2005–2012. Clinl Infect Dis. 2016;63(4):478–486. doi: 10.1093/cid/ciw248
- O’Grady K-AF, Kelpie L, Andrews RM, et al. The epidemiology of invasive group a streptococcal disease in Victoria, Australia. Med J Aust. 2007;186(11):565–569. doi: 10.5694/j.1326-5377.2007.tb01054.x
- O’Loughlin RE, Roberson A, Cieslak PR, et al. The epidemiology of invasive group a streptococcal infection and potential vaccine implications: United States, 2000-2004. Clinl Infect Dis. 2007;45(7):853–862. doi: 10.1086/521264
- Svensson N, Öberg S, Henriques B, et al. Invasive group a streptococcal infections in Sweden in 1994 and 1995: epidemiology and clinical spectrum. Scand J Infect Dis. 2000;32(6):609–614. doi: 10.1080/003655400459504
- Carapetis JR, Steer AC, Mulholland EK, et al. The global burden of group a streptococcal diseases. Lancet Infect Dis. 2005;5(11):685–694. doi: 10.1016/S1473-3099(05)70267-X
- Gherardi G, Vitali LA, Creti R. Prevalent emm types among invasive GAS in Europe and North America since year 2000. Front Public Health. 2018;6:59. doi: 10.3389/fpubh.2018.00059
- Luca-Harari B, Darenberg J, Neal S, et al. Clinical and microbiological characteristics of severe Streptococcus pyogenes disease in Europe. J Clin Microbiol. 2009;47(4):1155–1165. doi: 10.1128/JCM.02155-08
- Walker MJ, Hollands A, Sanderson-Smith ML, et al. Dnase Sda1 provides selection pressure for a switch to invasive group a streptococcal infection. Nature Med. 2007;13(8):981–985. doi: 10.1038/nm1612
- Hollands A, Pence MA, Timmer AM, et al. Genetic switch to hypervirulence reduces colonization phenotypes of the globally disseminated group a Streptococcus M1T1 clone. J Infect Dis. 2010;202(1):11–19. doi: 10.1086/653124
- Hassell M, Fagan P, Carson P, et al. Streptococcal necrotising fasciitis from diverse strains of Streptococcus pyogenes in tropical northern Australia: case series and comparison with the literature. BMC Infect Dis. 2004;4(1):60. doi: 10.1186/1471-2334-4-60
- Norton R, Smith HV, Wood N, et al. Invasive group a streptococcal disease in North Queensland (1996 – 2001). Indian J Med Res. 2004;119:148–151.
- Richardson LJ, Towers RJ, Cheng AC, et al. Diversity of emm sequence types in group a beta-haemolytic streptococci in two remote northern territory indigenous communities: implications for vaccine development. Vaccine. 2010;28(32):5301–5305. doi: 10.1016/j.vaccine.2010.05.046
- Maamary PG, Sanderson-Smith ML, Aziz RK, et al. Parameters governing invasive disease propensity of non-M1 serotype group a streptococci. J Innate Immun. 2010;2(6):596–606. doi: 10.1159/000317640
- Tsatsaronis JA, Ly D, Pupovac A, et al. Group a Streptococcus modulates host inflammation by manipulating polymorphonuclear leukocyte cell death responses. J Innate Immun. 2015;7:612–622. doi: 10.1159/000430498
- Kobayashi SD, Malachowa N, Deleo FR. Neutrophils and bacterial immune evasion. J Innate Immun. 2018;10(5–6):432–441. doi: 10.1159/000487756
- Edwards RJ, Pyzio M, Gierula M, et al. Proteomic analysis at the sites of clinical infection with invasive Streptococcus pyogenes. Sci Rep. 2018;8(1). doi: 10.1038/s41598-018-24216-2
- Kobayashi SD, Deleo FR. Role of neutrophils in innate immunity: a systems biology-level approach. Wiley Interdiscip Rev Syst Biol Med. 2009;1(3):309–333. doi: 10.1002/wsbm.32
- Kobayashi SD, Voyich JM, Buhl CL, et al. Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc Natl Acad Sci USA. 2002;99:6901–6906.
- Bratton DL, Henson PM. Neutrophil clearance: when the party is over, clean-up begins. Trends Immunol. 2011;32(8):350–357. doi: 10.1016/j.it.2011.04.009
- Kobayashi SD, Voyich JM, Braughton KR, et al. Down-regulation of proinflammatory capacity during apoptosis in human polymorphonuclear leukocytes. J Immunol. 2003b;170(6):3357–3368. doi: 10.4049/jimmunol.170.6.3357
- Lawrence SM, Corriden R, Nizet V. How neutrophils meet their end. Trends Immunol. 2020;41(6):531–544. doi: 10.1016/j.it.2020.03.008
- Liu L, Sun B. Neutrophil pyroptosis: new perspectives on sepsis. Cell Mol Life Sci. 2019;76(11):2031–2042. doi: 10.1007/s00018-019-03060-1
- Epstein FH, Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320(6):365–376. doi: 10.1056/NEJM198902093200606
- Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003;5(14):1317–1327. doi: 10.1016/j.micinf.2003.09.008
- Kobayashi SD, Braughton KR, Whitney AR, et al. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc Natl Acad Sci USA. 2003a;100:10948–10953.
- Williams JG, Ly D, Geraghty NJ, et al. Streptococcus pyogenes M1T1 variants induce an inflammatory neutrophil phenotype including activation of inflammatory caspases. Front Cell Infect Microbiol. 2021;10. doi: 10.3389/fcimb.2020.596023
- McLaughlin RE, Ferretti JJ. Electrotransformation of Streptococci. Methods Mol Biol. 1995;47:185–193 doi: 10.1385/0-89603-310-4:185
- Thomas HB, Moots RJ, Edwards SW, et al. Whose gene is it anyway? The effect of preparation purity on neutrophil transcriptome studies. PLoS One. 2015;10(9):e0138982. doi: 10.1371/journal.pone.0138982
- Flaherty RA, Puricelli JM, Higashi DL, et al. Streptolysin S promotes programmed cell death and enhances inflammatory signaling in epithelial keratinocytes during group a Streptococcus infection. Infect Immun. 2015;83(10):4118–4133. doi: 10.1128/IAI.00611-15
- Timmer AM, Timmer JC, Pence MA, et al. Streptolysin O promotes group a streptococcus immune evasion by accelerated macrophage apoptosis. J Biol Chem. 2009;284(2):862–871. doi: 10.1074/jbc.M804632200
- Nagata S, Suzuki J, Segawa K, et al. Exposure of phosphatidylserine on the cell surface. Cell Death Diff. 2016;23(6):952–961. doi: 10.1038/cdd.2016.7
- Daigle I, Simon HU. Critical role for caspases 3 and 8 in neutrophil but not eosinophil apoptosis. Int Arch Allergy Immunol. 2001;126(2):147–156. doi: 10.1159/000049506
- Miao EA, Leaf IA, Treuting PM, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11(12):1136–1142. doi: 10.1038/ni.1960
- Sollberger G, Strittmatter GE, Kistowska M, et al. Caspase-4 is required for activation of inflammasomes. J Immunol. 2012;188(4):1992–2000. doi: 10.4049/jimmunol.1101620
- Norrby-Teglund A, Chatellier S, Low DE, et al. Host variation in cytokine responses to superantigens determine the severity of invasive group a streptococcal infection. Eur J Immunol. 2000;30(11):3247–3255. doi: 10.1002/1521-4141(200011)30:11<3247:AID-IMMU3247>3.0.CO;2-D
- Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277(1):61–75. doi: 10.1111/imr.12534
- Arnaout MA. Structure and function of the leukocyte adhesion molecule CD11/CD18. Blood. 1990;75(5):1037–1050. doi: 10.1182/blood.V75.5.1037.1037
- Power C, Wang JH, Sookhai S, et al. Proinflammatory effects of bacterial lipoprotein on human neutrophil activation status, function and cytotoxic potential in vitro. Shock. 2001;15(6):461–466. doi: 10.1097/00024382-200115060-00009
- Skubitz KM, Campbell KD, Skubitz APN. CD66a, CD66b, CD66c, and CD66d each independently stimulate neutrophils. J Leukoc Biol. 1996;60(1):106–117. doi: 10.1002/jlb.60.1.106
- Dransfield I, Buckle AM, Savill JS, et al. Neutrophil apoptosis is associated with a reduction in CD16 (Fc gamma RIII) expression. J Immunol. 1994;153(3):1254–1263. doi: 10.4049/jimmunol.153.3.1254
- Fossati G, Moots RJ, Bucknall RC, et al. Differential role of neutrophil Fcγ receptor IIIb (CD16) in phagocytosis, bacterial killing, and responses to immune complexes. Arthritis & Rheumatism. 2002;46(5):1351–1361. doi: 10.1002/art.10230
- Brown S, Heinisch I, Ross E, et al. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature. 2002;418(6894):200–203. doi: 10.1038/nature00811
- Kurosaka K, Takahashi M, Watanabe N, et al. Silent cleanup of very early apoptotic Cells by macrophages. J Immunol. 2003;171(9):4672–4679. doi: 10.4049/jimmunol.171.9.4672
- Zeilhofer HU, Schorr W. Role of interleukin-8 in neutrophil signaling. Current Opinion Hematol. 2000;7(3):178–182. doi: 10.1097/00062752-200005000-00009
- Bréchard S, Bueb J-L, Tschirhart E. Interleukin-8 primes oxidative burst in neutrophil-like HL-60 through changes in cytosolic calcium. Cell Calcium. 2005;37(6):531–540. doi: 10.1016/j.ceca.2005.01.019
- McFarlin BK, Williams RR, Venable AS, et al. Image-based cytometry reveals three distinct subsets of activated granulocytes based on phagocytosis and oxidative burst. Cytometry Part A. 2013;83A(8):745–751. doi: 10.1002/cyto.a.22330
- Greenlee-Wacker MC, Rigby KM, Kobayashi SD, et al. Phagocytosis of staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J Immunol. 2014;192(10):4709–4717. doi: 10.4049/jimmunol.1302692
- Bergsbaken T, Cookson BT. Innate immune response during Yersinia infection: critical modulation of cell death mechanisms through phagocyte activation. J Leukoc Biol. 2009;86(5):1153–1158. doi: 10.1189/jlb.0309146
- Holzinger D, Gieldon L, Mysore V, et al. Staphylococcus aureus Panton‐Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J Leukoc Biol. 2012;92(5):1069–1081. doi: 10.1189/jlb.0112014
- Ryu JC, Kim MJ, Kwon Y, et al. Neutrophil pyroptosis mediates pathology of P. aeruginosa lung infection in the absence of the NADPH oxidase NOX2. Mucosal Immunol. 2017;10(3):757–774. doi: 10.1038/mi.2016.73
- Medina E, Goldmann O, Toppel AW, et al. Survival of Streptococcus pyogenes within host phagocytic cells: a pathogenic mechanism for persistence and systemic invasion. J Infect Dis. 2003a;187(4):597–603. doi: 10.1086/373998
- Medina E, Rohde M, Chhatwal GS. Intracellular survival of Streptococcus pyogenes in polymorphonuclear cells results in increased bacterial virulence. Infect Immun. 2003b;71(9):5376–5380. doi: 10.1128/IAI.71.9.5376-5380.2003