
ABSTRACT
Phenuiviruses are a class of segmented negative-sense single-stranded RNA viruses, typically consisting of three RNA segments that encode four distinct proteins. The emergence of pathogenic phenuivirus strains, such as Rift Valley fever phlebovirus (RVFV) in sub-Saharan Africa, Severe Fever with Thrombocytopenia Syndrome Virus (SFTSV) in East and Southeast Asia, and Heartland Virus (HRTV) in the United States has presented considerable challenges to global public health in recent years. The innate immune system plays a crucial role as the initial defense mechanism of the host against invading pathogens. In addition to continued research aimed at elucidating the epidemiological characteristics of phenuivirus, significant advancements have been made in investigating its viral virulence factors (glycoprotein, non-structural protein, and nucleoprotein) and potential host–pathogen interactions. Specifically, efforts have focused on understanding mechanisms of viral immune evasion, viral assembly and egress, and host immune networks involving immune cells, programmed cell death, inflammation, nucleic acid receptors, etc. Furthermore, a plethora of technological advancements, including metagenomics, metabolomics, single-cell transcriptomics, proteomics, gene editing, monoclonal antibodies, and vaccines, have been utilized to further our understanding of phenuivirus pathogenesis and host immune responses. Hence, this review aims to provide a comprehensive overview of the current understanding of the mechanisms of host recognition, viral immune evasion, and potential therapeutic approaches during human pathogenic phenuivirus infections focusing particularly on RVFV and SFTSV.
Phenuiviruses
The order Bunyavirales is one of the largest groups of RNA viruses with 636 members distributed across 14 families, including Arenaviridae, Cruliviridae, Discoviridae, Fimoviridae, Hantaviridae, Leishbuviridae, Mypovir-idae, Nairoviridae, Peribunyaviridae, Phasmaviridae, Phen-uiviridae, Tospoviridae, Tulasviridae, and Wupedeviridae [Citation1]. To date, six families of bunyaviruses (Arenaviridae, Peribunyaviridae, Nairoviridae, Phenuiviridae, Hanta-viridae, and Tulasviridae) have been reported to harbor human pathogenic viral strains. Phenuiviruses are one of the best-characterized bunyaviruses, with 20 genera and 137 species currently identified [Citation1]. These phenuiviruses are arthropod-borne viruses transmitted by ticks, sandflies, or mosquitoes, posing a serious public health threat worldwide. Examples include the Rift Valley fever virus (RVFV) and severe fever with thrombocytopenia syndrome virus (SFTSV) [Citation2–4], which are listed as severe infectious diseases with an urgent need for accelerated research and development by the World Health Organization Research and Development Blueprint [Citation5].
RVFV was reported in the Rift Valley of Kenya in 1931 and has since caused widespread epidemics in domesticated ruminants and humans in sub-Saharan Africa [Citation2,Citation3]. RVFV induces serious illness in ruminant animals, including sheep, goats, and cattle, leading to outbreaks of abortion and high mortality rates among neonatal lambs and calves [Citation6]. Since the 1950s, there has been an increase in the scope and frequency of RVFV outbreaks, with notable epidemics occurring throughout Africa. By the early 2000s, RVFV had spread beyond Africa to the Arabian Peninsula, with reported cases in Yemen and Saudi Arabia [Citation7,Citation8]. Human infections primarily occur through direct contact with blood, body fluids, or tissues of infected animals, rather than through mosquito-borne transmission [Citation9]. The clinical manifestations of RVFV infection in humans can vary from mild flu-like symptoms to severe conditions, such as hemorrhagic fever [Citation10–12].
SFTSV is an emerging tick-borne phenuivirus that was first reported in China in 2010 [Citation4,Citation13–15], causing a severe viral hemorrhagic fever termed SFTS with a case fatality ranging from 16% to 30% in East and Southeast Asia, including South Korea, Japan, Thailand, Vietnam, and Pakistan [Citation4,Citation16–20]. In addition, the vector for SFTSV, Haemaphysalis longicornis, is widely distributed in Australia and New Zealand and has more recently been identified in the United States. This suggests that the geographic range of SFTSV may extend beyond Asia [Citation21], further highlighting the public health significance of SFTSV. The primary clinical features in SFTS patients include fever, thrombocytopenia, leukocytopenia, vomiting, diarrhea, hemorrhage, and even multiple organ failure [Citation4]. Notably, it has been observed that SFTSV can be sporadically transmitted through oral and ocular membranes via contact with body fluids of SFTS patients [Citation22–24].
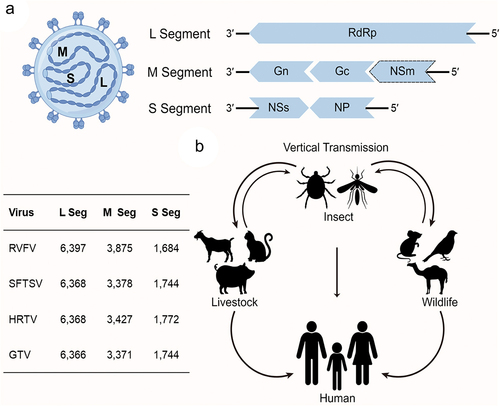
As the largest family of Bunyavirales, Phenuiviridae contains numerous members that can infect three kingdoms, including animals, plants, and fungi [Citation1]. Phenuiviruses are a group of cytoplasmically enveloped, icosahedral spherical RNA viruses, with an average envelope diameter ranging from 80 to 160 nm. Phenuivirus virions contain two to eight single-stranded RNA segments, but most species that infect vertebrates have three RNA fragments, including the large (L), medium (M), and small (S) RNA segments (). Each genomic segment exhibits a pseudo-circular and panhandle-like structure due to the complementary of terminal sequence. The L segment encodes the RNA-dependent RNA polymerase (RdRp), which is responsible for the transcription and replication of the phenuivirus genome after it is released into the cytoplasm. The M segment encodes the glycoprotein precursor, which is cleaved into Gn and Gc by host proteases in the endoplasmic reticulum (ER) or Golgi apparatus. The glycoprotein is responsible for the assembly of the phenuivirus into a lipid envelope and for the entry of the phenuivirus virion into its target cell. Of note, some phenuiviruses, such as RVFV, encode a non-structural (NSm) protein at the N-terminus of the glycoprotein precursor. The S segment encodes proteins in an ambisense strategy, which covers two open reading frames and encodes non-structural (NSs) protein by positive-sense and nucleoprotein (NP) by negative-sense, respectively. The NSs protein serves as an important virulence factor, while NP is the most abundant phenuivirus protein that interacts with RdRp and phenuivirus genomic RNA to form the ribonucleoprotein (RNP) complex, which is the basic replication unit of phenuivirus. Phenuiviruses lack internal matrix components, and the phenuivirus NP plays an important role in the interaction and protection of the phenuivirus genomic RNA. Of note, the N arm of NP could be targeted by Moloney murine leukemia virus 10 protein and thereby blocks RNP formation by inhibiting NP polymerization, NP-RNA binding, and NP-RdRp interaction processes [Citation25].
Figure 1. (a) Schematic diagram of phenuivirus structure and (b) transmission cycle of phenuivirus. (a) Phenuvirus comprises three RNA segments large (L) segment, medium (M) segment, and small (S) segment, which encodes RNA-dependent RNA polymerase (RdRp), glycoprotein, non-structural (NS) protein, and nucleoprotein (NP), respectively. The L and M segments are encoded in a negative-sense orientation, whereas the S segment is encoded in an ambisense orientation. Additionally, the table in the left notes the length of each segment for RVFV, SFTSV, HRTV, and GTV.
The life cycle of a phenuivirus can be divided into several phases: binding, internalization, replication, assembly, and exocytosis [Citation26,Citation27]. Cell surface proteins, lipids, and carbohydrates can all function as phenuivirus entry receptors, promoting virus uptake through receptor-mediated endocytosis or phagocytosis. To date, many cell surface receptors, such as low-density lipoprotein receptor-related protein 1, C-type lectins (DC-SIGN, L-SIGN, LSECtin), heparan sulfates, nonmuscle myosin heavy-chain type IIA (NMMHC-IIA), C-C motif chemokine receptor 2, and sorting nexin 11 have been identified to facilitate the entry of phenuiviruses [Citation28–33]. Of note, phenuivirus transport is a highly dynamic biological process [Citation34]. Following the binding of phenuivirus glycoproteins to target cell surface receptors, phenuiviruses are transported via clathrin- or caveolin-mediated endocytosis, micropinocytosis, or other unknown mechanisms into Rab5a-positive early endosomes [Citation27,Citation35–37]. Early endosomes acquire a lower pH environment and become Rab7a-positive and lysosomal-associated membrane protein 1 (LAMP-1) positive late endosomes [Citation35]. The acidic environment in late endosomes induces a conformational change in phenuivirus glycoproteins, further triggering fusion of the phenuivirus envelope with the endosomal membrane and resulting in the release of phenuivirus RNPs into the cytoplasm. Released RNPs serve as templates for primary phenuivirus transcription and are translocated to the rough ER, where they promote viral replication, transcription, and protein synthesis [Citation34,Citation38]. Like all segmented negative-sense RNA viruses, phenuiviruses also employ cap snatching for viral replication in the cytoplasm [Citation39]. The RdRp of phenuivirus is responsible for cap binding and cleavage of host-derived capped mRNA fragments (10–25 nucleotides downstream of the 5' cap that lack a poly(A) tail), which serve as primers for viral transcription. WNT-CTNNB1 signaling pathway-related RNAs might be the source of SFTSV cap-snatching [Citation40]. Additionally, translation of phenuiviruses at the ER is accompanied by the cleavage of glycoproteins into Gn and Gc, which are then transported to the Golgi apparatus. Notably, Golgi-specific brefeldin A-resistance guanine nucleotide exchange factor 1 (GBF1), residing at the cis-Golgi crucial for Golgi-ER trafficking, plays a significant role in the assembly of the phenuvirus Uukuniemi virus (UUKV) through its interaction with Gn and Gc [Citation41]. This interaction allows phenuiviruses, including RVFV, UUKV, and Toscana virus (TOSV), to exploit GBF1 for enhanced infectivity [Citation41]. To form progeny phenuivirus virions, the assembled phenuivirus RNP complex is integrated into the lumen of the ER-Golgi intermediate compartment (ERGIC) or Golgi apparatus through interaction with the cytoplasmic tail of the glycoprotein Gn. Mature phenuivirus virions are eventually released into the extracellular environment via secretory vesicles [Citation26,Citation42].
Given the increasing frequency and number of phenuivirus outbreaks, phenuiviruses pose a serious threat to global public health [Citation5]. However, no specific vaccines or drugs have been approved against human pathogenic phenuiviruses, in part due to the lack of a comprehensive understanding of the interaction between phenuiviruses and the innate immune system [Citation43]. The aim of this review is to highlight antiviral innate immunity and viral immune escape mechanisms during human pathogenic phenuivirus infections, focusing particularly on RVFV and SFTSV. By addressing and uncovering these potential links, we aim not only to enhance our understanding of intricate phenuivirus–host interactions but also to contribute to the development of potential antiviral therapies.
Recognition of phenuiviruses by cytoplasmic nucleic acid receptors
The innate immune response constitutes the first line of host defense against infection by various viruses (). Phenuivirus replication depends on primer-independent RNA synthesis, indicating that phenuiviruses with 5'-triphosphate genomic RNAs can trigger retinoic acid-inducible gene-I (RIG-I)-dependent innate immunity [Citation44–51]. It is noteworthy that RIG-I can also recognize packaged RNPs [Citation46]. Apart from RIG-I, targeting melanoma differentiation-associated gene 5 (MDA5) cannot prevent the induction of type I interferons (IFNs) induced by RVFV genomic RNAs, suggesting that MDA5 is not the major PRR in response to phenuivirus infections [Citation50]. Interestingly, MDA5 deficiency in dendritic cells reduces the induction of type I IFNs in the early phase of RVFV infection [Citation49], and MDA5 is able to recognize SFTSV infection, albeit to a much lesser extent [Citation44]. This can be explained by the accumulation of mitochondrial double-stranded RNA (dsRNA) [Citation52,Citation53] or unreported pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) in the cytoplasm during phenuivirus infections, which needs further investigation. It is worth noting that phenuiviruses and other negative-stranded bunyaviruses do not produce substantial amounts of dsRNA, as no detectable amounts of dsRNA are found in cells infected by hantavirus, LaCrosse virus, and Akabane virus [Citation54–56]. This phenomenon can be elucidated by the genome replication strategy employed by bunyaviruses, wherein genomic and anti-genomic RNA molecules are encapsidated by viral nucleoprotein during synthesis. This process likely impedes the formation of extended regions of dsRNA by hindering the complementary base pairing of the genomic and anti-genomic RNA strands [Citation57].
Figure 2. Phenuivirus protein components antagonize the pattern recognition receptor induced antiviral effect. Phenuivirus structure or non-structure protein can directly target multiple antiviral proteins for immune escape, including cGAS, STING, TRIM25, MAVS, TBK1, etc.
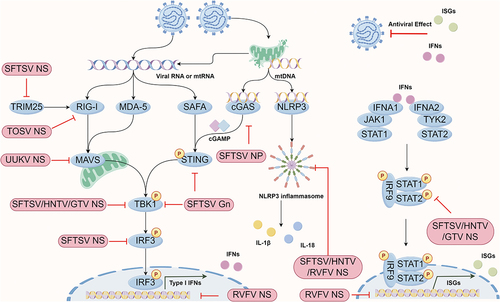
Additionally, during RVFV infection, endosomal Toll-like receptors (TLRs, including TLR3, TLR7, and TLR8), as well as TLR adaptor molecule, do not promote the production of type I IFNs and inflammasome-related genes [Citation49,Citation58]. As for SFTSV, TLR7 and TLR8 deficiency significantly inhibit NF-κB pathway activation, whereas TLR7 but not TLR8 deficiency inhibits SFTSV replication in THP-1 cells, a monocyte-derived macrophage [Citation59]. Furthermore, MAVS and MyD88 are required for the induction of type I IFNs and inflammatory cytokines, emphasizing the importance of RLRs and TLRs in anti-SFTSV immune responses [Citation51].
Nuclear heterogeneous nuclear ribonucleoproteins (hnRNPs) are a class of RNA chaperones involved in RNA biology and transcriptional regulatory network. Importantly, a large number of hnRNPs are involved in viral infection through different mechanisms, including DNA viruses, RNA viruses, and retroviruses [Citation60]. The nuclear scaffold attachment factor A (SAFA), also known as hnRNPU, was identified in 2019 as a nuclear dsRNA sensor in a cytoplasm-independent manner [Citation61]. SAFA exhibits an antiviral role in restricting the replication of multiple viruses, including HSV-1, VSV, and HIV-1 [Citation61,Citation62]. Interestingly, SAFA functions as an RNA PRR in restricting the phenuivirus SFTSV infection in macrophages and embryonic stem cells [Citation63]. Under SFTSV infection conditions, SAFA is retained in the cytoplasm and recognizes cytoplasmic SFTSV RNA. SAFA then recruits and activates the STING-TBK1 axis-dependent type I IFN signaling to restrict SFTSV infection. Notably, SAFA-mediated SFTSV recognition can be divided into three stages: (1) SAFA directly recognizes SFTSV NP via its nuclear localization signal (NLS) motif. Similar results are observed with RVFV and Heartland virus (HRTV) NP; (2) SAFA recognizes cytoplasmic SFTSV genomic RNAs; (3) Activated SAFA interacts with STING and promotes STING-TBK1-dependent antiviral signaling [Citation63]. Moreover, SFTSV NSs can also interact with SAFA and mediate the retention of SAFA in the cytoplasm [Citation63], suggesting that the NSs may capture SAFA into IBs to mediate the suppression of SAFA.
Like RNA sensors, DNA sensors are also crucial for inducing the type I IFN-mediated antiviral response upon infection with RNA viruses. Numerous studies have highlighted the role of the cGAS-STING pathway in RNA virus recognition, primarily dependent on mislocalized mitochondrial DNA (mtDNA). Interestingly, the DNA sensor cGAS also participates in recognizing the phenuivirus SFTSV infection by detecting mtDNA released into the cytoplasm [Citation59,Citation64,Citation65]. As a countermeasure, SFTSV employs NP and Gn, rather than NSs, to suppress the cGAS-STING-mediated antiviral response. NP and Gn interact with cGAS and STING, respectively, facilitating their degradation through autophagy [Citation64]. Notably, SFTSV Gn interacts with STING to prevent its dimerization and inhibit K27-linked ubiquitination of STING, thereby disrupting the assembly of the STING-TBK1 complex [Citation65].
The NSs protein of phenuiviruses evading antiviral type I IFN responses
Phenuiviruses have evolved multiple countermeasures against the activation of innate immunity (). The NSs protein of phenuivirus is considered as the most important virulence factor, as NSs proteins in phenuiviruses and other Bunyavirales consistently suppress the host immune response, despite their sequence diversity [Citation66]. The NSs protein of RVFV was the first identified and is one of the best-known types I IFN antagonists among phenuiviruses until the emergence of the highly pathogenic SFTSV. Unlike most NSs proteins of phenuiviruses, the NSs protein of RVFV can not only be present in the cytoplasm but also localize and form a unique filamentous structure (depending on the ΩXaV motif) in the nucleus (depending on the N-terminal PXXP motif) [Citation67–69]. RVFV lacking NSs is unable to inhibit the IFN response and is non-pathogenic in the mouse model [Citation70,Citation71]. Importantly, the antagonistic activity of the RVFV NSs protein is, in part, due to its localization in the nucleus [Citation72–74]. Notably, a recent study resolved the crystal structure of a truncated form of the RVFV NSs core domain (residues 83–248) [Citation75]. The recombinant RVFV encoding the NSs core domain induces intranuclear filaments, suggesting that neither the truncated N-terminal nor the C-terminal NSs regions are required for fibrillation [Citation75]. Mechanistically, the RVFV NSs protein disrupts type I interferons via multiple ways, which not only induces transcriptional shutoff via inhibiting the RNA polymerase II preinitiation complex transcription factor II H (TFIIH) assembly but also acts as an E3 ligase complex, promoting the proteasomal degradation of the TFIIH p62 subunit and the dsRNA-dependent protein kinase (PKR) [Citation76–81]. In addition, the NSs protein of RVFV prevents type I IFN production by specifically binding to the SAP30-YY1 complex and blocking nuclear mRNA export [Citation81–84]. Importantly, a double cysteine-to-serine substitution at residues 39 and 40 of the RVFV NSs protein abolishes its antagonistic effect on type I IFN induction and reduces degradation of PKR [Citation85]. Like RVFV, the NSs protein of TOSV can also target PKR and RIG-I, promoting proteasome-dependent degradation [Citation86,Citation87]. Interestingly, the TOSV NSs protein exhibits E3 ubiquitin ligase activity localized to both the carboxy- and amino-terminal domains, which facilitates the ubiquitination of RIG-I, ultimately promoting its degradation [Citation86,Citation88].
The SFTSV NSs protein has the ability to form inclusion bodies (IBs, also known as viroplasm-like structures (VLSs)) in the cytoplasm, which interferes with RIG-I- and MDA5-mediated antiviral responses, facilitating immune escape by trapping downstream molecules within the IBs, while the presence of the PXXP motif at its N-terminus is crucial for IB formation and NSs-dependent antagonist activity [Citation89]. Currently, various IFN-related molecules, including TRIM25, RIG-I, TBK1, IKKε, IRF3, IRF7, STAT1, STAT2, and LSm14A [Citation44,Citation66,Citation89–96], as well as the cell cycle regulation protein CDK1 [Citation97], have been reported to be sequestered into IBs by the SFTSV NSs protein to dampen the innate immune response. Remarkably, recent research has revealed that the SFTSV NSs protein-induced IBs are autophagic or autophagy-like vesicles [Citation98]. Specifically, the NSs protein facilitates the sequestration of antiviral proteins, such as TBK1, into autophagic vesicles via interaction with LC3 for degradation and evasion of antiviral immune responses. This process is mediated by the induction of complete autophagy through the promotion of Beclin-1-dependent autophagy initiation complex formation [Citation98]. Additionally, the SFTSV NSs protein has been shown to sequester mTOR into IBs [Citation99] or attenuated the inhibitory effect of vimentin on Beclin1 [Citation100], thereby inducing autophagy.
Genetically closely related to SFTSV, HRTV causes acute febrile illness in humans [Citation4,Citation101]. HRTV infection inhibits the phosphorylation and translocation of STAT2 and STAT1, thereby impeding antiviral responses [Citation102]. Notably, the HRTV NSs protein diffuses in the cytoplasm and acts as a potent antagonist, efficiently preventing the activation of TBK1 and STAT2 [Citation66,Citation102,Citation103]. This antagonistic effect may be dependent on the PXXP motif [Citation89]. Additionally, the HRTV NSs protein can interact with STAT1, but to a lesser extent than with STAT2. However, it cannot interfere with the phosphorylation and translocation of STAT1, suggesting that the HRTV NSs protein cannot inhibit type II IFN signaling [Citation66]. UUKV is apathogenic and not associated with human diseases [Citation104]. The NSs protein of UUKV diffuses into the cytoplasm and weakly antagonizes the human IFN-β promoter activation compared to those of SFTSV and HRTV [Citation66]. Mechanistically, the UUKV NSs protein captures MAVS and thus inhibits the induction of type I IFNs [Citation66,Citation105,Citation106]. The Guertu virus (GTV) is a potentially pathogenic phenuivirus reported in China [Citation107]. The GTV NSs protein is capable of forming IBs and extensive filamentous structures (FSs) in the cytoplasm [Citation108], which is unique among phenuiviruses. The GTV NSs protein sequesters TBK1 and STAT2 into viral IBs and FSs, thereby weakening antiviral innate immunity [Citation108]. Further exploration is needed to determine whether GTV NSs-induced IBs and FSs have novel functions in GTV pathogenicity.
The complex role of NLRP3 inflammasome in phenuivirus infections
Inflammatory responses are complex biological processes essential in the initial phase of innate immunity against virus infections, while excessive inflammation responses could be associated with fatal infection outcome during virus infections. NLRP3 is the most well-characterized inflammasome associated with RNA virus challenges, playing a pivotal role in eliciting inflammatory responses [Citation109,Citation110]. The role of NLRP3 has been implicated in recognizing RVFV and Hantaan virus (HTNV) infections [Citation58,Citation111]. RVFV barely activates the NLRP3 inflammasome compared to the RVFV NSs-deficient strain, suggesting the involvement of RVFV NSs in impairing NLRP3 recognition [Citation58]. The priming process induced by LPS is necessary for NLRP3 inflammasome activation during RVFV infection but not for UV-inactivated RVFV. Additionally, NLRP3, ASC, and caspase-1 deficiency almost completely block IL-1β production, indicating that RVFV-induced inflammasome activation primarily depends on NLRP3, while the absence of RLRs or MyD88 does not affect inflammasome activation during RVFV infection. Regarding HTNV, NLRP3 inflammasome activation induced by HTNV relies on ROS release but not extracellular ATP [Citation111,Citation112]. Transcriptomic and quantitative analyses have highlighted the tight association between inflammatory responses and the pathogenicity of SFTSV [Citation59,Citation113,Citation114]. SFTSV infection, but not UV-inactivated SFTSV, induces IL-1β production in THP-1 cell and human peripheral blood mononuclear cell monocyte, but not lymphocyte, primarily through the NLRP3 inflammasome [Citation59,Citation115,Citation116]. The TLR7/8-MyD88 axis, rather than MAVS, is crucial in the NLRP3 priming phase during SFTSV infection by triggering NF-κB activation [Citation51,Citation59]. Mechanistically, cytosolic oxidized mtDNA binds and triggers NLRP3 inflammasome activation [Citation59,Citation64,Citation65], while NLRP3 stimulators calcium influx and potassium efflux do not contribute to the induction of IL-1β during SFTSV infection [Citation115].
It is worth noting that the current understanding of the role of SFTSV NSs in NLRP3 and inflammatory response activation seems controversial. On the one hand, both SFTSV and HRTV NSs can impair the activation of the NLRP3 inflammasome in HEK293T cells [Citation89], and the substitution of alanine at positions 20 to 23 of SFTSV/HRTV NSs partially blocks the activation of NLRP3 inflammasome. As for inflammatory responses, the SFTSV NSs protein targets ABIN2, thus promoting the activation of the TPL2/ABIN2/p105 complex, which induces the production of the anti-inflammatory cytokine IL-10, dampening innate immunity and promoting viral pathogenesis [Citation117]. Notably, SFTSV-NSs P102A and K211R mutants are unable to promote IL-10 production via the TPL2 signaling pathway [Citation117]. NRF2 plays a pivotal role in the antioxidant signaling pathway, ultimately contributing to anti-inflammatory responses [Citation118]. The SFTSV NSs protein binds to the SPRY domain of TRIM21 via the K226KTDG230 motif and inhibits the function of TRIM21[Citation119]. Subsequently, the SFTSV NSs protein promotes p62 oligomerization and p62-mediated Keap1 sequestration, thereby inducing the dissociation of NRF2 from Keap1 to activate the antioxidant signaling pathway. This results in the expression of CD36 and lipid uptake, contributing to the replication and pathogenesis of SFTSV. These studies suggest that the NSs-TRIM21 interaction is essential for the pathogenesis of SFTSV [Citation119]. Notably, SFTSV NSs mutant (K226KTDG230) can still modulate the induction of IFN and activation of IL-10 signaling pathway [Citation119].
On the other hand, the SFTSV life cycle depends on NLRP3 activation in microglial cells, as knockout or inhibition of NLRP3 significantly suppresses SFTSV replication and attenuates SFTSV-induced cell death [Citation120]. The N-terminal fragment of NSs interacts with NLRP3 to promote the formation of inflammasome complex, thereby promoting the replication of SFTSV through an unclear mechanism. In addition, overexpression of SFTSV components (RdRp, Gn/Gc, NP, or NSs) does not prevent LPS-induced IL-1β production in macrophages [Citation115]. Such contradictory results suggest that NLRP3 inflammasome plays an important part in SFTSV infection, but their complex interrelationships require further investigation. In addition, the overexpression of NSs can barely induce the production of inflammatory cytokines, whereas NSs can enhance the activation of NF-κB-induced by SeV infection or TNF-α treatment. Notably, NSs disrupts the interaction between TBK1 and IKKβ, thereby promoting the activation of NF-κB-mediate inflammatory responses [Citation121]. Treatment with the NF-κB inhibitor, SC75741, reduces viral RNA synthesis of SFTSV and HRTV and inhibits SFTSV infection [Citation122]. Moreover, p38 plays crucial roles in inflammatory responses, while p38 inhibition by SB203580 effectively inhibits the replication of SFTSV [Citation123].
Phenuiviruses exploit autophagy as a platform for virus assembly and egress
Autophagy is a complex process during viral infection that plays a multifaceted role in host defense. It can protect the host from viral infection by degrading invading viruses, enhancing antigen presentation, or promoting both inflammatory and non-inflammatory responses [Citation124–126]. However, despite its classical role in limiting viral infection, autophagy can sometimes be exploited by viruses to provide a platform for replication and exocytosis, especially if viruses can survive in the lysosome or autolysosome or evade lysosomal degradation [Citation127–129].
To date, limited studies have investigated the involvement of autophagy in the life cycle of phenuiviruses. The TLR7-MyD88 axis, but not other canonical TLRs, has been identified as crucial for mediating the autophagy flux during RVFV infection, facilitated by the direct interaction between RVFV glycoprotein and TLR7 [Citation130]. Various canonical autophagy processes, from preinitiation to elongation, have been demonstrated to be essential in restricting RVFV infection [Citation130]. However, contrasting findings suggest that RVFV-NP induces autophagy to facilitate RVFV replication in macrophages [Citation131]. Similarly, recent studies have investigated the interplay between autophagy and SFTSV. Analysis of transcriptional and protein patterns has confirmed the involvement of Beclin-1-dependent classical autophagy flux during SFTSV infection [Citation42,Citation132]. Notably, SFTSV infection promotes the accumulation of p62, a phenomenon induced by SFTSV NSs interaction with TRIM21, thereby enhancing the stability and oligomerization of p62 [Citation119]. In addition, novel roles of SFTSV components, including Gn, NSs, and NP, in inducing autophagy have been identified [Citation42,Citation65,Citation99,Citation100] (). Activation of UPR has been linked to the stimulation of the autophagy process. Furthermore, activation of the unfolded protein response (UPR) has been observed during infection with SFTSV, HTNV, and Tula hantavirus [Citation133–135]. The UPR appears to play a beneficial role in promoting SFTSV replication, with specific involvement of the SFTSV glycoprotein (GP) in UPR activation [Citation133].
Figure 3. (a) Schematic presentation of the phenuivirus replication cycle and (b) autophagy induction pattern of phenuivirus. (a) Phenuivirus glycoprotein (GP) is synthesized by membrane-bound ribosomes at the ER and subsequently translocated to the ERGIC and Golgi complex. GP is anticipated to recruit the RNP complex for viral assembly and maturation. Subsequently, mature virus particles are released from the cell via secretory vesicles or autophagic vesicles. Alternatively, CD63-positive exosomes also facilitate the exocytosis of phenuivirus in a receptor-independent manner. (b) SFTSV NP induces classical autophagy by disrupting the association between beclin-1 and BCL2. SFTSV NSs induces classical autophagy through several mechanisms, including sequestering mTOR into IBs, promoting vimentin degradation via K48-linked ubiquitin-proteasome pathway, or facilitating the formation of beclin-1-dependent autophagy initiation complexes. Notably, IBs induced by SFTSV NSs may function as autophagy-like vesicles that sequester antiviral proteins such as TBK1, thereby promoting TBK1 degradation for immune evasion. Additionally, RVFV NP interacts with p62 and LC3 to induce autophagy and subsequently inhibit the antiviral innate immune response.
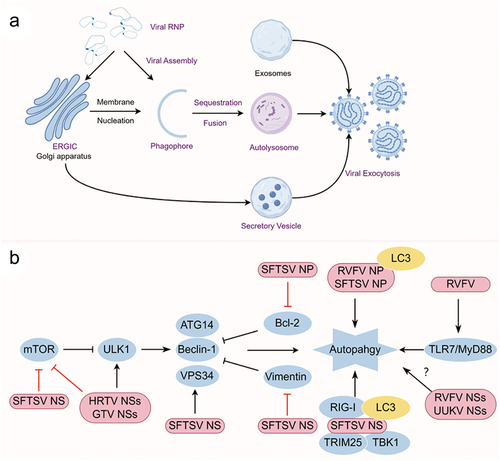
To date, numerous studies have investigated the processes of internalization, replication, recruitment, and assembly of bunyaviruses [Citation26,Citation27]. Bunyavirus RNPs are typically considered to be recruited to the lumen of the Golgi apparatus for assembly and egress by budding into Golgi-mediated secretory vesicles [Citation26]. However, recent research has unveiled that SFTSV manipulates the autophagy flux and exploits autophagy for assembly and egress [Citation42]. Under SFTSV infection, the activity of lysosomal cathepsin B and D enzymes is reduced, while lysosomal pH remains unchanged. This reduction in enzyme activity is accompanied by the localization of SFTSV particles in autophagic vesicles, which are utilized for exocytosis. Lipidated LC3, PI3KC3, and ATG12-ATG5-ATG16L1 complexes, rather than the ULK1 complex, are detected in purified SFTSV particles. In addition, the ERGIC and Golgi apparatus, but not the ER, serve as the primary membrane sources for autophagosome formation and act as platforms for SFTSV assembly. These novel findings present a new pattern of bunyavirus assembly and egress, where autophagosomes originating from the ERGIC and/or Golgi apparatus serve as SFTSV assembly platforms, and the fusion of autophagy-dependent vesicles with lysosomes is crucial for SFTSV exocytosis. In addition to the autophagic vesicles, CD63-positive extracellular vesicles could also facilitate the transmission of SFTSV and RVFV in a receptor-independent manner [Citation136,Citation137]. These observations pave the way for potential SFTSV treatments by targeting the transmission of the virus ().
Apoptosis is beneficial for the replication of phenuivirus RVFV and SFTSV
Apoptosis is a highly regulated form of cell death, characterized by membrane blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, DNA fragmentation, and mRNA decay [Citation138]. RVFV infection triggers apoptosis via caspase-8, while NSm expression suppresses caspase-8 activation [Citation139]. The C-terminal region of the RVFV NSm targets the mitochondrial outer membrane and exerts an anti-apoptotic role [Citation140]. During RVFV infection, p53 phosphorylation is induced and is dependent on the RVFV NSs but not NSm [Citation141,Citation142]. Importantly, p53 localizes to the nucleus and partially co-localizes with RVFV NSs during RVFV infection. Loss of p53 reduces the replication of RVFV and RVFV-induced cell death, presumably by manipulating the p53-dependent apoptosis signal pathway [Citation141,Citation143]. In addition, RVFV triggers cell apoptosis in astrocytes, whereas the RVFV NSs protein may hinder apoptosis by sequestering activated caspase-3 within the nucleus [Citation144,Citation145]. During SFTSV infection, apoptosis may be appropriately repressed or minimally enhanced in monocytes [Citation59,Citation146], whereas severe apoptosis is triggered in peripheral monocytes from SFTS patient at the early stage of fatal SFTSV infection [Citation147]. Additionally, apoptosis is induced in mDCs, HepG2 cells, and endothelial cells during SFTSV infection [Citation147–149]. Further proteomic analysis reveals that a cluster of anti-apoptosis proteins is induced during SFTSV infection, such as SOD2, BCL3, CD74, FAM129B [Citation59], indicating that SFTSV could partially block the activation of apoptosis. Notably, inhibition of apoptosis by PS-341 can block the replication of SFTSV in 293T and Vero cells [Citation150].
Phenuivirus infections and adaptive immune responses
Dendritic cells (DCs) have been identified as target cells for multiple phenuiviruses, such as RVFV and UUKV [Citation151]. Lethally infected monkeys have more monocytes and plasmacytoid dendritic cells (pDCs) in peripheral blood mononuclear cells (PBMCs), while the numbers of myeloid dendritic cells (mDCs) fluctuated during infection but did not differ significantly between fatal and survivor groups [Citation152]. In addition, a heightened presence of circulating mDCs is associated with a protective effect in patients SFTS [Citation153]. Specifically, the proportions of both mDCs and pDCs in PBMCs are significantly reduced in patients during the acute phase of SFTS, particularly in those who succumbed to the illness, while the reduction in pDCs is comparable between patients who survived and those who did not. Intriguingly, during the recovery phase, the levels of circulating mDCs and pDCs return to similar levels as those of healthy individuals [Citation153]. Importantly, the decreased percentage of DCs in PBMCs was correlated with cytokine levels, viral load, and the severity of SFTS [Citation153,Citation154]. The down-regulation of CD80/CD86 and TLR3 on mDCs of SFTS patients could be the underlying mechanism [Citation153,Citation154].
Elevated Natural killer (NK) cells and overexpression of ISGs and genes related to NK cell functions suggests that these cells may play an active role in controlling brain infection during RVFV infection [Citation155,Citation156]. Research on patients with SFTS consistently demonstrates a notable reduction in the proportion of NK cells in peripheral blood during the initial stages of SFTSV infection, with this reduction being inversely correlated with the severity of the disease [Citation157–159]. Notably, an increased presence of granulocytic myeloid-derived suppressor cells (gMDSCs) in SFTS patients highlights their role in inhibiting the antiviral response of NK cells [Citation159]. However, conflicting findings have also been reported, suggesting that SFTS patients may exhibit a higher percentage of NK cells in peripheral blood [Citation160,Citation161].
The importance of adaptive immunity in controlling RVFV infection was studied in C57BL/6J mice via infection with RVFV lacking NSs [Citation162]. Notably, the depletion of B cells, CD4+ T cells, and CD8+ T cells revealed that B cells and CD4+ T cells played crucial roles in viral clearance, while CD8+ T cells not. Particularly noteworthy is the finding that depletion of CD4+ T cells leads to elevated viral RNA levels, decreased antibody titers, and stronger proinflammatory responses, underscoring the essential role of CD4+ T cells in generating robust neutralizing antibody and proinflammatory responses associated with RVFV clearance [Citation162]. As for SFTSV, numerous studies have reported a significant increase in B cell levels in individuals with SFTS, with a notably higher percentage of B cells observed in fatal cases compared to mild cases [Citation161,Citation163–165]. Notably, the capacity of B cells to generate specific antibodies is significantly impaired. On the one hand, SFTSV infection not only hinders the development, antigen presentation, and intercellular communication of B cells but also impedes B cell class-switching process [Citation157,Citation166]. Patients with SFTS in the acute phase show a decrease in CD3+ T cells compared to healthy controls and convalescent patients, with a significant reduction in deceased patients [Citation157,Citation161,Citation167,Citation168]. Additionally, there is also a negative association between CD4+ T cell quantity and SFTS severity, but no correlation with CD8+ T cells [Citation161,Citation167]. Research on CD4+ T cell subsets in SFTS patients indicates elevated percentages of Th1, Th2, Th17 cells during the acute phase, with an increase in Th1 cells during recovery. Conversely, Treg cells decreased in acute phase compared to convalescent patients and healthy controls, indicating a possible link between Th1, Th2, Treg cell numbers, and disease severity in SFTS patients [Citation167]. Furthermore, SFTSV infection induces a downregulation of arginine, which hampers the virus clearance function of T cells by impairing the CD3-ζ chain of T cells [Citation169].
Gut microbiota and platelet in phenuivirus infection
The increasing prevalence of emerging infectious viral diseases underscores the importance of investigating the gut microbiota’s involvement and therapeutic possibilities. Recent research using 16S rRNA-sequencing has provided insight into the potential correlation between gut microbiota and patients with SFTS [Citation170,Citation171]. The probiotic Akkermansia muciniphila was found to be more prevalent in surviving patients compared to deceased individuals with SFTS [Citation171], which could help create targeted microbiota treatments for people at risk of severe infections. Mechanistically, A. muciniphila and its metabolite harmaline have been shown to specifically upregulate bile acid-CoA: amino acid N-acyltransferase in hepatocytes. Consequently, elevated levels of chenodeoxycholic acid (GCDCA and TCDCA) result in the inhibition of TGR5-NF-κB-mediated systemic inflammatory responses, ultimately contributing to the mitigation of the severity and mortality rates associated with SFTS. It is noteworthy that the vector gut microbiota may serve as a potential target for mitigating viral infections [Citation172]. The current research demonstrates that RVFV or SFTSV infection significantly impacts the primary vectors of microbial diversity, richness, and composition [Citation173,Citation174], variations in microbiota may play a role in the infection and transmission dynamics of phenuiviruses.
Viral hemorrhagic fevers are caused by bunyaviruses, including RVFV, SFTSV, Crimean-Congo hemorrhagic fever virus (CCHFV), and a number of hantaviruses, leading to systemic symptoms like fever and low platelet count [Citation4,Citation10–12,Citation175]. Of note, thrombocytopenia stands out as a mortality risk factor strongly associated with the severity of SFTS, exerting significant impacts on SFTS deterioration and fatal outcomes [Citation176]. Recent studies have shed light on the effective internalization and replication of SFTSV within platelets [Citation177,Citation178]. SFTSV interacts with circulating platelets via the Gn/Gc-platelet glycoprotein VI interaction, leading to platelet activation and apoptosis characterized by increased P-selectin expression and phosphatidylserine exposure on the platelet surface, respectively. Activated platelets are predisposed to aggregate and adhere to endothelial cells, thereby promoting platelet clearance through enhanced platelet-macrophage crosstalk. Moreover, SFTSV or SFTSV Gn can directly induce platelet activation and apoptosis in a ROS-MAPK-dependent manner, which can be effectively attenuated by the administration of antioxidants.
Conclusions and opening questions
The global spread of phenuiviruses presents significant public health risks, as the intricate viral-mediated immune responses remain poorly understood. Efforts to elucidate the detailed mechanisms of phenuivirus interactions with host defense are in their nascent stages. This review explores the established role and underlying mechanisms of innate immunity in combating SFTSV infection while also identifying unresolved questions and critical gaps that require further investigation.
1) While recent research has broadened our understanding of the interplay between phenuivirus and innate immunity, including autophagy, inflammasome, and novel PRR, there remains a significant gap in our knowledge of phenuivirus pathogenicity. This gap has hindered the progress in developing effective therapeutics for phenuivirus-related illnesses. 2) To avoid the recognition of host, phenuiviruses have developed multiple immune escape mechanisms via blocking or hijacking cellular responses directly or indirectly for the viral life cycle during coevolution. Hence, a comprehensive understanding of virus-host protein interactions is to some content essential for the development of antiviral therapies. 3) Different phenuivirus NSs proteins exhibit relatively low amino acid similarity, and the structure and cellular distribution of NSs proteins are also highly specific. Notably, some NSs proteins of phenuiviruses are non-cytotoxic, and some Hantavirus HNTV lacks NSs proteins, indicating that other phenuivirus or even bunyavirus components could have cooperated or even replaced the function of NSs protein in disturbing the activation of innate immunity for viral life cycle. 4) Autophagy may play a key role in bunyavirus assembly and release, with viral components like Gn, NP, and NSs triggering autophagy. These components are likely to work together during infection to use autophagy for various viral processes. Investigating the specific mechanisms involved in these processes is essential for advancing our knowledge of bunyavirus biology.
Author contributions statement
ZZJ, CMZ, and NL organized and wrote the main manuscript text; XJY and CMZ reviewed the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Additional information
Funding
References
- Kuhn JH, Adkins S, Agwanda BR, et al. Taxonomic update of phylum negarnaviricota (Riboviria: Orthornavirae), including the large orders bunyavirales and Mononegavirales. Arch Virol. 2021 Dec;166(12):3513–17. doi: 10.1007/s00705-021-05143-6.
- Daubney R, Hudson J, Garnham P. Enzootic hepatitis or rift valley fever: an undescribed virus disease of sheep, cattle, and man from East Africa. Pathol And Bacteriol. 1931;34(4):545–579. doi: 10.1002/path.1700340418
- Tigoi C, Sang R, Chepkorir E, et al. High risk for human exposure to rift valley fever virus in communities living along livestock movement routes: a cross-sectional survey in Kenya. PLOS Negl Trop Dis. 2020;14(2):e0007979. doi: 10.1371/journal.pntd.0007979
- Yu XJ, Liang MF, Zhang SY, et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med. 2011;364(16):1523–1532. doi: 10.1056/NEJMoa1010095
- 2018 annual review of diseases prioritized under the research and development blueprint. WHO Research and Development Blueprint; 2018. Available from: http://www.who.int/emergencies/diseases/2018prioritization-report.pdf
- Bird BH, Githinji JW, Macharia JM, et al. Multiple virus lineages sharing recent common ancestry were associated with a large rift valley fever outbreak among livestock in Kenya during 2006-2007. J Virol. 2008;82(22):11152–11166. doi: 10.1128/JVI.01519-08
- Madani TA, Al-Mazrou YY, Al-Jeffri MH, et al. Rift Valley fever epidemic in Saudi Arabia: epidemiological, clinical, and laboratory characteristics. Clin Infect Dis. 2003;37(8):1084–1092. doi: 10.1086/378747
- Centers for Disease C, Prevention. Outbreak of Rift Valley fever–Saudi Arabia, August-October, MMWR Morb Mortal Wkly Rep. 2000;49(40):905–908.
- Nicholas DE, Jacobsen KH, Waters NM. Risk factors associated with human rift valley fever infection: systematic review and meta-analysis. Trop Med Int Health. 2014;19(12):1420–1429. doi: 10.1111/tmi.12385
- Ikegami T, Makino S. The pathogenesis of rift valley fever. Viruses. 2011;3(5):493–519. doi: 10.3390/v3050493
- Wilson LR, McElroy AK. Rift valley fever virus encephalitis: viral and host determinants of pathogenesis. Annu Rev Virol. 2024. doi: 10.1146/annurev-virology-093022-011544
- Ebogo-Belobo JT, Kenmoe S, Abanda NN, et al. Contemporary epidemiological data of Rift Valley fever virus in humans, mosquitoes and other animal species in Africa: a systematic review and meta-analysis. Vet Med Sci. 2023;9(5):2309–2328. doi: 10.1002/vms3.1238
- Sato Y, Mekata H, Sudaryatma PE, et al. Isolation of severe fever with thrombocytopenia syndrome virus from various tick species in area with human severe fever with thrombocytopenia syndrome cases. Vector-Borne Zoonot. 2021;21(5):378–384. doi: 10.1089/vbz.2020.2720
- Luo LM, Zhao L, Wen HL, et al. Haemaphysalis longicornis ticks as reservoir and vector of severe fever with thrombocytopenia syndrome virus in China. Emerg Infect Dis. 2015;21(10):1770–1776. doi: 10.3201/eid2110.150126
- Fang LZ, Xiao X, Lei SC, et al. Ticks as a competent vector of severe fever with thrombocytopenia syndrome virus. Ticks Tick-Borne Dis. 2023;14(2):14. doi: 10.1016/j.ttbdis.2022.102100
- Kim KH, Yi J, Kim G, et al. Severe fever with thrombocytopenia syndrome, South Korea, Emerg Infect Dis 2013; 2012;19(11):1892–1894. doi: 10.3201/eid1911.130792
- Takahashi T, Maeda K, Suzuki T, et al. The first identification and retrospective study of severe fever with thrombocytopenia syndrome in Japan. J Infect Dis. 2014;209(6):816–827. doi: 10.1093/infdis/jit603
- Tran XC, Yun Y, Van an L, et al. Endemic severe fever with thrombocytopenia syndrome, Vietnam. Emerg Infect Dis. 2019;25(5):1029–1031. doi: 10.3201/eid2505.181463
- Ongkittikul S, Watanawong R, Rompho P. Severe fever with thrombocytopenia syndrome virus: the first case report in Thailand. BKK Med J. 2020 Sep 25;16(2):204. doi: 10.31524/bkkmedj.2020.22.001
- Zohaib A, Zhang J, Saqib M, et al. Serologic evidence of severe fever with thrombocytopenia syndrome virus and related viruses in Pakistan. Emerg Infect Dis. 2020;26(7):1513–1516. doi: 10.3201/eid2607.190611
- Rainey T, Occi JL, Robbins RG, et al. Discovery of haemaphysalis longicornis (Ixodida: Ixodidae) parasitizing a sheep in New Jersey, United States. J Med Entomol. 2018;55(3):757–759. doi: 10.1093/jme/tjy006
- Liu Y, Li Q, Hu WF, et al. Person-to-person transmission of severe fever with thrombocytopenia syndrome virus. Vector-Borne Zoonot. 2012;12(2):156–160. doi: 10.1089/vbz.2011.0758
- Bao CJ, Qi X, Wang H. A novel bunyavirus in China. N Engl J Med. 2011;365:862–863.
- Zhou C-M, Qi R, Qin X-R, et al. Oral and ocular transmission of severe fever with thrombocytopenia syndrome virus. Infect Med. 2022;1(1):2–6. doi: 10.1016/j.imj.2021.12.002
- Mo Q, Xu Z, Deng F, et al. Host restriction of emerging high-pathogenic bunyaviruses via MOV10 by targeting viral nucleoprotein and blocking ribonucleoprotein assembly. PLOS Pathog. 2020;16(12):e1009129. doi: 10.1371/journal.ppat.1009129
- Wichgers Schreur PJ, Kortekaas J, Palese P. Single-molecule FISH reveals non-selective packaging of rift valley fever virus genome segments. PLOS Pathog. 2016;12(8):e1005800. doi: 10.1371/journal.ppat.1005800
- Liu J, Xu M, Tang B, et al. Single-particle tracking reveals the sequential entry process of the bunyavirus severe fever with thrombocytopenia syndrome virus. Small. 2019;15(6):e1803788. doi: 10.1002/smll.201803788
- Koch J, Xin Q, Tischler ND, et al. Entry of phenuiviruses into mammalian Host cells. Viruses. 2021;13(2):299. doi: 10.3390/v13020299
- Albornoz A, Hoffmann AB, Lozach PY, et al. Early bunyavirus-host cell interactions. Viruses. 2016;8(5):143. doi: 10.3390/v8050143
- Schwarz MM, Price DA, Ganaie SS, et al. Oropouche orthobunyavirus infection is mediated by the cellular host factor Lrp1. Proc Natl Acad Sci USA. 2022;119(33):e2204706119. doi: 10.1073/pnas.2204706119
- Ganaie SS, Schwarz MM, McMillen CM, et al. Lrp1 is a host entry factor for rift valley fever virus. Cell. 2021;184(20):5163–78 e24. doi: 10.1016/j.cell.2021.09.001
- Zhang L, Peng X, Wang Q, et al. CCR2 is a host entry receptor for severe fever with thrombocytopenia syndrome virus. Sci Adv. 2023;9(31):eadg6856. doi: 10.1126/sciadv.adg6856
- Liu T, Li J, Liu Y, et al. SNX11 identified as an essential Host factor for SFTS virus infection by CRISPR knockout screening. Virol Sin. 2019;34(5):508–520. doi: 10.1007/s12250-019-00141-0
- Sun Y, Li J, Gao GF, et al. Bunyavirales ribonucleoproteins: the viral replication and transcription machinery. Crit Rev Microbiol. 2018;44(5):522–540. doi: 10.1080/1040841X.2018.1446901
- Lozach PY, Mancini R, Bitto D, et al. Entry of bunyaviruses into mammalian cells. Cell Host & Microbe. 2010;7(6):488–499. doi: 10.1016/j.chom.2010.05.007
- Garrison AR, Radoshitzky SR, Kota KP, et al. Crimean–Congo hemorrhagic fever virus utilizes a clathrin- and early endosome-dependent entry pathway. Virology. 2013;444(1–2):45–54. doi: 10.1016/j.virol.2013.05.030
- Harmon B, Schudel BR, Maar D, et al. Rift valley fever virus strain MP-12 enters mammalian host cells via caveola-mediated endocytosis. J Virol. 2012;86(23):12954–12970. doi: 10.1128/JVI.02242-12
- Shi X, van Mierlo Jt, French A, et al. Visualizing the replication cycle of bunyamwera orthobunyavirus expressing fluorescent protein-tagged gc glycoprotein. J Virol. 2010;84(17):8460–8469. doi: 10.1128/JVI.00902-10
- Olschewski S, Cusack S, Rosenthal M. The cap-snatching mechanism of bunyaviruses. Trends Microbiol. 2020;28(4):293–303. doi: 10.1016/j.tim.2019.12.006
- Jiang XM, Xin QL, Liu K, et al. Regulation of the WNT-CTNNB1 signaling pathway by severe fever with thrombocytopenia syndrome virus in a cap-snatching manner. MBio. 2023;14(6):e0168823. doi: 10.1128/mbio.01688-23
- Uckeley ZM, Moeller R, Kuhn LI, et al. Quantitative proteomics of uukuniemi virus-host cell interactions reveals GBF1 as proviral host factor for phleboviruses. Mol Cell Proteomics. 2019;18(12):2401–2417. doi: 10.1074/mcp.RA119.001631
- Yan JM, Zhang WK, Yan LN, et al. Bunyavirus SFTSV exploits autophagic flux for viral assembly and egress. Autophagy. 2022;18(7):1–14. doi: 10.1080/15548627.2021.1994296
- Zhou CM, Yu XJ. Unraveling the underlying interaction mechanism between dabie bandavirus and innate immune response. Front Immunol. 2021;12:676861. doi: 10.3389/fimmu.2021.676861
- Min YQ, Ning YJ, Wang H, et al. A RIG-I–like receptor directs antiviral responses to a bunyavirus and is antagonized by virus-induced blockade of TRIM25-mediated ubiquitination. J Biol Chem. 2020;295(28):9691–9711. doi: 10.1074/jbc.RA120.013973
- Mukherjee P, Woods TA, Moore RA, et al. Activation of the innate signaling molecule MAVS by bunyavirus infection upregulates the adaptor protein SARM1, leading to neuronal death. Immunity. 2013;38(4):705–716. doi: 10.1016/j.immuni.2013.02.013
- Weber M, Gawanbacht A, Habjan M, et al. Incoming RNA virus nucleocapsids containing a 5'-triphosphorylated genome activate rig-I and Antiviral signaling. Cell Host Microbe. 2013;13(3):336–346. doi: 10.1016/j.chom.2013.01.012
- Spengler JR, Patel JR, Chakrabarti AK, et al. RIG-I mediates an antiviral response to Crimean-Congo hemorrhagic fever virus. J Virol. 2015;89(20):10219–10229. doi: 10.1128/JVI.01643-15
- Huang C, Kolokoltsova OA, Yun NE, et al. Junín virus infection activates the type I interferon pathway in a RIG-I-Dependent manner. PLOS Negl Trop Dis. 2012;6(5):e1659. doi: 10.1371/journal.pntd.0001659
- Ermler ME, Yerukhim E, Schriewer J, et al. RNA helicase signaling is critical for type i interferon production and protection against rift valley fever virus during mucosal challenge. J Virol. 2013;87(9):4846–4860. doi: 10.1128/JVI.01997-12
- Habjan M, Andersson I, Klingstrom J, et al. Processing of genome 5' termini as a strategy of negative-strand RNA viruses to avoid RIG-I-Dependent interferon induction. PLOS ONE. 2008;3(4):e2032. doi: 10.1371/journal.pone.0002032
- Yamada S, Shimojima M, Narita R, et al. RIG-I-Like receptor and toll-like receptor signaling pathways cause aberrant production of inflammatory Cytokines/Chemokines in a severe fever with thrombocytopenia syndrome virus infection mouse Model. J Virol. 2018;92(13). doi: 10.1128/JVI.02246-17
- Wu X, Qi X, Liang M, et al. Roles of viroplasm-like structures formed by nonstructural protein NSs in infection with severe fever with thrombocytopenia syndrome virus. FASEB J. 2014;28(6):2504–2516. doi: 10.1096/fj.13-243857
- Dhir A, Dhir S, Borowski LS, et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. 2018;560(7717):238–242. doi: 10.1038/s41586-018-0363-0
- Weber F, Wagner V, Rasmussen SB, et al. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol. 2006;80(10):5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006
- Wang H, Vaheri A, Weber F, et al. Old world hantaviruses do not produce detectable amounts of dsRNA in infected cells and the 5' termini of their genomic RNAs are monophosphorylated. Journal Of General Virology. 2011;92(5):1199–1204. doi: 10.1099/vir.0.029405-0
- O’Brien CA, Hobson-Peters J, Yam AW, et al. Viral RNA intermediates as targets for detection and discovery of novel and emerging mosquito-borne viruses. PLOS Negl Trop Dis. 2015;9(3):e0003629. doi: 10.1371/journal.pntd.0003629
- Barr JN. Bunyavirus mRNA synthesis is coupled to translation to prevent premature transcription termination. RNA. 2007;13(5):731–736. doi: 10.1261/rna.436607
- Ermler ME, Traylor Z, Patel K, et al. Rift valley fever virus infection induces activation of the NLRP3 inflammasome. Virology. 2014;449:174–180. doi: 10.1016/j.virol.2013.11.015
- Li S, Li H, Zhang YL, et al. SFTSV infection induces BAK/BAX-Dependent mitochondrial DNA release to trigger NLRP3 Inflammasome activation. Cell Reports. 2020;30(13):4370–85 e7. doi: 10.1016/j.celrep.2020.02.105
- Wan Q, Song D, Li H, et al. Stress proteins: the biological functions in virus infection, present and challenges for target-based antiviral drug development. Signal transduction and targeted therapy. Signal Transduct Target Ther. 2020;5(1):125. doi: 10.1038/s41392-020-00233-4
- Cao L, Liu S, Li Y, et al. The nuclear matrix protein SAFA surveils viral RNA and Facilitates immunity by activating antiviral enhancers and super-enhancers. Cell Host & Microbe. 2019;26:369–84 e8. doi: 10.1016/j.chom.2019.08.010
- Valente ST, Goff SP. Inhibition of HIV-1 gene expression by a fragment of hnRNP U. Mol Cell. 2006;23(4):597–605. doi: 10.1016/j.molcel.2006.07.021
- Liu BY, Yu XJ, Zhou CM, et al. SAFA initiates innate immunity against cytoplasmic RNA virus SFTSV infection. PLOS Pathog. 2021;17(11):e1010070. doi: 10.1371/journal.ppat.1010070
- Zz J, Chu M, Ln Y, et al. SFTSV nucleoprotein mediates DNA sensor cGAS degradation to suppress cGAS-dependent antiviral responses. Microbiol Spectr. 2024;12(6):e0379623. doi: 10.1128/spectrum.03796-23
- Jia Y, Li F, Liu Z, et al. Interaction between the SFTSV envelope glycoprotein gn and STING inhibits the formation of the STING-TBK1 complex and suppresses the nf-κB signaling pathway. J Virol. 2024;98(3):e0181523. doi: 10.1128/jvi.01815-23
- Rezelj L VV, Chaudhary P, Elliott V, et al. Differential antagonism of human innate immune responses by tick-borne phlebovirus nonstructural proteins. mSphere. 2017;2(3). doi: 10.1128/mSphere.00234-17
- Yadani FZ, Kohl A, Prehaud C, et al. The carboxy-terminal acidic domain of rift valley fever virus NSs protein is essential for the formation of filamentous structures but not for the nuclear localization of the protein. J Virol. 1999;73(6):5018–5025. doi: 10.1128/JVI.73.6.5018-5025.1999
- Billecocq A, Spiegel M, Vialat P, et al. NSs protein of rift valley fever virus blocks interferon production by inhibiting host gene transcription. J Virol. 2004;78(18):9798–9806. doi: 10.1128/JVI.78.18.9798-9806.2004
- Cyr N, de la Fuente C, Lecoq L, et al. A OmegaXaV motif in the Rift Valley fever virus NSs protein is essential for degrading p62, forming nuclear filaments and virulence. Proc Natl Acad Sci U S A. 2015 May 12;112(19):6021–6026. doi: 10.1073/pnas.1503688112.
- Bouloy M, Janzen C, Vialat P, et al. Genetic evidence for an interferon-antagonistic function of rift valley fever virus nonstructural protein NSs. J Virol. 2001;75(3):1371–1377. doi: 10.1128/JVI.75.3.1371-1377.2001
- Vialat P, Billecocq A, Kohl A, et al. The S segment of rift valley fever phlebovirus (bunyaviridae) carries determinants for attenuation and virulence in mice. J Virol. 2000;74(3):1538–1543. doi: 10.1128/JVI.74.3.1538-1543.2000
- Leger P, Nachman E, Richter K, et al. NSs amyloid formation is associated with the virulence of rift valley fever virus in mice. Nat Commun. 2020;11(1):3281. doi: 10.1038/s41467-020-17101-y
- Ly HJ, Ikegami T. Rift valley fever virus NSs protein functions and the similarity to other bunyavirus NSs proteins. Virol J. 2016;13(1):118. doi: 10.1186/s12985-016-0573-8
- Wuerth JD, Weber F. Phleboviruses and the type I interferon response. Viruses. 2016;8(6):174. doi: 10.3390/v8060174
- Barski M, Brennan B, Miller OK, et al. Rift valley fever phlebovirus NSs protein core domain structure suggests molecular basis for nuclear filaments. Elife. 2017;6:6. doi: 10.7554/eLife.29236
- Kalveram B, Lihoradova O, Ikegami T. NSs protein of rift valley fever virus promotes posttranslational downregulation of the TFIIH subunit p62. J Virol. 2011;85(13):6234–6243. doi: 10.1128/JVI.02255-10
- Habjan M, Pichlmair A, Elliott RM, et al. NSs protein of rift valley fever virus induces the specific degradation of the double-stranded RNA-dependent protein kinase. J Virol. 2009;83(9):4365–4375. doi: 10.1128/JVI.02148-08
- Ikegami T, Narayanan K, Won S, et al. Rift valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and Inhibits eIf2α phosphorylation. PLOS Pathog. 2009;5(2):e1000287. doi: 10.1371/journal.ppat.1000287
- Kainulainen M, Lau S, Samuel CE, et al. NSs virulence factor of rift valley fever virus engages the F-Box proteins FBXW11 and β-TRCP1 to degrade the antiviral protein kinase PKR. J Virol. 2016;90(13):6140–6147. doi: 10.1128/JVI.00016-16
- Mudhasani R, Tran JP, Retterer C, et al. Protein kinase R degradation is essential for rift valley fever virus infection and is regulated by SKP1-CUL1-F-box (SCF)FBXW11-NSs E3 ligase. PLOS Pathog. 2016;12(2):e1005437. doi: 10.1371/journal.ppat.1005437
- Terasaki K, Ramirez SI, Makino S, et al. Mechanistic insight into the host transcription inhibition function of rift valley fever virus NSs and its importance in virulence. PLOS neglected tropical diseases. PLOS Negl Trop Dis. 2016;10(10):e0005047. doi: 10.1371/journal.pntd.0005047
- Weill L, Shestakova E, Bonnefoy E. Transcription factor YY1 binds to the murine beta interferon promoter and regulates its transcriptional capacity with a dual activator/repressor role. J Virol. 2003;77(5):2903–2914. doi: 10.1128/JVI.77.5.2903-2914.2003
- Le May N, Mansuroglu Z, Leger P, et al. A SAP30 complex inhibits ifn-β expression in rift valley fever virus infected cells. PLOS Pathog. 2008;4(1):e13. doi: 10.1371/journal.ppat.0040013
- Copeland AM, Van Deusen NM, Schmaljohn CS. Rift valley fever virus NSS gene expression correlates with a defect in nuclear mRNA export. Virology. 2015;486:88–93. doi: 10.1016/j.virol.2015.09.003
- Monteiro GER, Jansen van Vuren P, Wichgers Schreur PJ, et al. Mutation of adjacent cysteine residues in the NSs protein of Rift Valley fever virus results in loss of virulence in mice. Virus Res. 2018;249:31–44. doi: 10.1016/j.virusres.2018.03.005
- Gori-Savellini G, Valentini M, Cusi MG. Toscana virus NSs protein inhibits the induction of type I interferon by interacting with RIG-I. J Virol. 2013;87(12):6660–6667. doi: 10.1128/JVI.03129-12
- Kalveram B, Ikegami T. Toscana virus NSs protein promotes degradation of double-stranded RNA-dependent protein kinase. J Virol. 2013;87(7):3710–3718. doi: 10.1128/JVI.02506-12
- Gori Savellini G, Anichini G, Gandolfo C, et al. Toscana virus non-structural protein NSs acts as E3 ubiquitin ligase promoting RIG-I degradation. PLOS Pathog. 2019;15(12):e1008186. doi: 10.1371/journal.ppat.1008186
- Moriyama M, Igarashi M, Koshiba T, et al. Two conserved amino acids within the NSs of severe fever with thrombocytopenia syndrome phlebovirus are essential for anti-interferon activity. J Virol. 2018;92(19). doi: 10.1128/JVI.00706-18
- Santiago FW, Covaleda LM, Sanchez-Aparicio MT, et al. Hijacking of RIG-I signaling proteins into virus-induced cytoplasmic structures correlates with the inhibition of type I interferon responses. J Virol. 2014;88(8):4572–4585. doi: 10.1128/JVI.03021-13
- Ning YJ, Wang M, Deng M, et al. Viral suppression of innate immunity via spatial isolation of TBK1/IKKε from mitochondrial antiviral platform. Journal Of Molecular Cell Biology. 2014;6(4):324–337. doi: 10.1093/jmcb/mju015
- Wu X, Qi X, Qu B, et al. Evasion of antiviral immunity through sequestering of TBK1/IKKε/IRF3 into viral inclusion bodies. J Virol. 2014;88(6):3067–3076. doi: 10.1128/JVI.03510-13
- Hong Y, Bai M, Qi X, et al. Suppression of the ifn-α and -β induction through sequestering IRF7 into viral inclusion bodies by nonstructural protein NSs in severe fever with thrombocytopenia syndrome bunyavirus infection. The Journal Of Immunology. 2019;202(3):841–856. doi: 10.4049/jimmunol.1800576
- Ning YJ, Feng K, Min YQ, et al. Disruption of type I interferon signaling by the nonstructural protein of severe fever with thrombocytopenia syndrome virus via the hijacking of STAT2 and STAT1 into inclusion bodies. J Virol. 2015;89(8):4227–4236. doi: 10.1128/JVI.00154-15
- Chaudhary V, Zhang S, Yuen KS, et al. Suppression of type I and type III IFN signalling by NSs protein of severe fever with thrombocytopenia syndrome virus through inhibition of STAT1 phosphorylation and activation. J Gen Virol. 2015;96(11):3204–3211. doi: 10.1099/jgv.0.000280
- Zhang L, Fu Y, Zhang R, et al. Nonstructural protein NSs hampers cellular antiviral response through LSm14A during severe fever with thrombocytopenia syndrome virus infection. The J Immunol. 2021;207(2):590–601. doi: 10.4049/jimmunol.2100148
- Liu S, Liu H, Kang J, et al. The severe fever with thrombocytopenia syndrome virus NSs protein interacts with CDK1 to induce G2 cell cycle arrest and positively regulate viral replication. J Virol. 2020;94(6). doi: 10.1128/JVI.01575-19
- Li ZM, Duan SH, Yu TM, et al. Bunyavirus SFTSV NSs utilizes autophagy to escape the antiviral innate immune response. Autophagy. 2024:1–13. doi: 10.1080/15548627.2024.2356505
- Feng K, Zhang H, Jiang Z, et al. SFTS bunyavirus NSs protein sequestrates mTOR into inclusion bodies and deregulates mTOR-ULK1 signaling, provoking pro-viral autophagy. J Med Virol. 2023;95(1):e28371. doi: 10.1002/jmv.28371
- Liu S, Su Y, Lu Z, et al. The SFTSV nonstructural proteins induce autophagy to promote viral replication via interaction with Vimentin. J Virol. 2023;97(4):e0030223. doi: 10.1128/jvi.00302-23
- McMullan LK, Folk SM, Kelly AJ, et al. A new phlebovirus associated with severe febrile illness in Missouri. N Engl J Med. 2012;367(9):834–841. doi: 10.1056/NEJMoa1203378
- Feng K, Deng F, Hu Z, et al. Heartland virus antagonizes type I and III interferon antiviral signaling by inhibiting phosphorylation and nuclear translocation of STAT2 and STAT1. J Biol Chem. 2019;294(24):9503–9517. doi: 10.1074/jbc.RA118.006563
- Ning YJ, Feng K, Min YQ, et al. Heartland virus NSs protein disrupts host defenses by blocking the TBK1 kinase–IRF3 transcription factor interaction and signaling required for interferon induction. Journal Of Biological Chemistry. 2017;292(40):16722–16733. doi: 10.1074/jbc.M117.805127
- Saikku P. Arboviruses in Finland. 3. Uukuniemi virus antibodies in human, cattle, and reindeer sera. Am J Trop Med Hyg. 1973;22(3):400–403. doi: 10.4269/ajtmh.1973.22.400
- Rezelj O VV, Elliott AK, Elliott, et al. Generation of mutant uukuniemi viruses lacking the nonstructural protein NSs by reverse genetics indicates that NSs is a weak interferon antagonist. J Virol. 2015;89(9):4849–4856. doi: 10.1128/JVI.03511-14
- Simons JF, Hellman U, Pettersson RF. Uukuniemi virus S RNA segment: ambisense coding strategy, packaging of complementary strands into virions, and homology to members of the genus phlebovirus. J Virol. 1990;64(1):247–255. doi: 10.1128/jvi.64.1.247-255.1990
- Shen S, Duan X, Wang B, et al. A novel tick-borne phlebovirus, closely related to severe fever with thrombocytopenia syndrome virus and heartland virus, is a potential pathogen. Emerging Microbes & Infections. 2018;7:95. doi: 10.1038/s41426-018-0093-2
- Min YQ, Shi C, Yao T, et al. The nonstructural protein of guertu virus disrupts Host defenses by blocking antiviral interferon induction and action. ACS Infect Dis. 2020;6(5):857–870. doi: 10.1021/acsinfecdis.9b00492
- Zhao C, Zhao W. NLRP3 inflammasome-A key player in antiviral responses. Front Immunol. 2020;11:211. doi: 10.3389/fimmu.2020.00211
- Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489. doi: 10.1038/s41577-019-0165-0
- Ye W, Lei Y, Yu M, et al. NLRP3 inflammasome is responsible for hantavirus inducing interleukin-1β in THP-1 cells. International Journal Of Molecular Medicine. 2015;35(6):1633–1640. doi: 10.3892/ijmm.2015.2162
- Ma R, Zhang X, Shu J, et al. Nlrc3 knockout mice showed renal pathological changes after HTNV infection. Front Immunol. 2021;12:692509. doi: 10.3389/fimmu.2021.692509
- Sun Y, Jin C, Zhan F, et al. Host cytokine storm is associated with disease severity of severe fever with thrombocytopenia syndrome. J Infect Dis. 2012;206(7):1085–1094. doi: 10.1093/infdis/jis452
- Liu MM, Lei XY, Yu H, et al. Correlation of cytokine level with the severity of severe fever with thrombocytopenia syndrome. Virol J. 2017;14(1):6. doi: 10.1186/s12985-016-0677-1
- Liu JW, Chu M, Jiao YJ, et al. SFTSV infection induced Interleukin-1beta secretion through NLRP3 Inflammasome activation. Front Immunol. 2021;12:595140. doi: 10.3389/fimmu.2021.595140
- Li Z, Hu J, Bao C, et al. Activation of the NLRP3 inflammasome and elevation of interleukin-1β secretion in infection by sever fever with thrombocytopenia syndrome virus. Sci Rep. 2022;12(1):2573. doi: 10.1038/s41598-022-06229-0
- Choi Y, Park SJ, Sun Y, et al. Severe fever with thrombocytopenia syndrome phlebovirus non-structural protein activates TPL2 signalling pathway for viral immunopathogenesis. Nat Microbiol. 2019;4(3):429–437. doi: 10.1038/s41564-018-0329-x
- Saha S, Buttari B, Panieri E, et al. An overview of Nrf2 signaling pathway and its role in inflammation. Molecules. 2020;25(22):25. doi: 10.3390/molecules25225474
- Choi Y, Jiang Z, Shin WJ, et al. Severe fever with thrombocytopenia syndrome virus NSs interacts with TRIM21 to activate the p62-Keap1-Nrf2 pathway. J Virol. 2020;94(6). doi: 10.1128/JVI.01684-19
- Gao C, Yu Y, Wen C, et al. Nonstructural protein NSs activates inflammasome and pyroptosis through interaction with NLRP3 in human microglial cells infected with severe fever with thrombocytopenia syndrome bandavirus. J Virol. 2022;96(13):e0016722. doi: 10.1128/jvi.00167-22
- Khalil J, Yamada S, Tsukamoto Y, et al. The nonstructural protein NSs of severe fever with thrombocytopenia syndrome virus causes a Cytokine storm through the hyperactivation of nf-κB. Mol Cell Biol. 2020;41(3). doi: 10.1128/MCB.00542-20
- Mendoza CA, Yamaoka S, Tsuda Y, et al. The nf-kappaB inhibitor, SC75741, is a novel antiviral against emerging tick-borne bandaviruses. Antiviral Res. 2021;185:104993. doi: 10.1016/j.antiviral.2020.104993
- Cheng Y, Sun F, Wang L, et al. Virus-induced p38 MAPK activation facilitates viral infection. Theranostics. 2020;10(26):12223–12240. doi: 10.7150/thno.50992
- Lee HK, Lund JM, Ramanathan B, et al. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315(5817):1398–1401. doi: 10.1126/science.1136880
- Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–206. doi: 10.1038/nature11866
- Orvedahl A, MacPherson S, Sumpter R Jr., et al. Autophagy protects against sindbis virus infection of the central nervous system. Cell Host & Microbe. 2010;7(2):115–127. doi: 10.1016/j.chom.2010.01.007
- Chen YH, Du W, Hagemeijer MC, et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell. 2015;160(4):619–630. doi: 10.1016/j.cell.2015.01.032
- Corona AK, Saulsbery HM, Velazquez AFC, et al. Enteroviruses remodel Autophagic trafficking through regulation of Host SNARE proteins to promote virus replication and cell exit. Cell Rep. 2018;22(12):3304–3314. doi: 10.1016/j.celrep.2018.03.003
- Ghosh S, Dellibovi-Ragheb TA, Kerviel A, et al. β-coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell. 2020;183(6):1520–1535.e14. doi: 10.1016/j.cell.2020.10.039
- Moy RH, Gold B, Molleston JM, et al. Antiviral autophagy restrictsRift valley fever virus infection and is conserved from flies to mammals. Immunity. 2014;40(1):51–65. doi: 10.1016/j.immuni.2013.10.020
- Zhu X, Guan Z, Fang Y, et al. Rift valley fever virus nucleoprotein triggers autophagy to dampen antiviral innate immune responses. J Virol. 2023;97(4):e0181422. doi: 10.1128/jvi.01814-22
- Yan JM, Zhang WK, Li F, et al. Integrated transcriptome profiling in THP-1 macrophages infected with bunyavirus SFTSV. Virus Res. 2021;306:198594. doi: 10.1016/j.virusres.2021.198594
- Zhang LK, Wang B, Xin Q, et al. Quantitative proteomic analysis reveals unfolded-protein response involved in severe fever with thrombocytopenia syndrome virus infection. J Virol. 2019;93(10). doi: 10.1128/JVI.00308-19
- Li Z, Shen Y, Song Y, et al. ER stress-related molecules induced by Hantaan virus infection in differentiated THP-1 cells. Cell Stress & Chaperones. 2021;26(1):41–50. doi: 10.1007/s12192-020-01150-9
- Li XD, Lankinen H, Putkuri N, et al. Tula hantavirus triggers pro-apoptotic signals of ER stress in Vero E6 cells. Virology. 2005;333(1):180–189. doi: 10.1016/j.virol.2005.01.002
- Silvas JA, Popov VL, Paulucci-Holthauzen A, et al. Extracellular vesicles mediate receptor-independent transmission of novel tick-borne bunyavirus. J Virol. 2016;90(2):873–886. doi: 10.1128/JVI.02490-15
- Ahsan NA, Sampey GC, Lepene B, et al. Presence of viral RNA and Proteins in exosomes from cellular clones resistant to rift valley fever virus infection. Front Microbiol. 2016;7:139. doi: 10.3389/fmicb.2016.00139
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–257. doi: 10.1038/bjc.1972.33
- Won S, Ikegami T, Peters CJ, et al. Nsm protein of rift valley fever virus suppresses virus-induced apoptosis. J Virol. 2007;81(24):13335–13345. doi: 10.1128/JVI.01238-07
- Terasaki K, Won S, Makino S. The C-terminal region of Rift Valley fever virus NSm protein targets the protein to the mitochondrial outer membrane and exerts antiapoptotic function. J Virol. 2013;87(1):676–682. doi: 10.1128/JVI.02192-12
- Austin D, Baer A, Lundberg L, et al. p53 activation following rift valley fever virus infection contributes to cell death and viral production. PLOS ONE. 2012;7(5):e36327. doi: 10.1371/journal.pone.0036327
- Popova TG, Turell MJ, Espina V, et al. Reverse-phase phosphoproteome analysis of signaling pathways induced by rift valley fever virus in human small airway epithelial cells. PLOS ONE. 2010;5(11):e13805. doi: 10.1371/journal.pone.0013805
- Narayanan A, Amaya M, Voss K, et al. Reactive oxygen species activate NFkappaB (p65) and p53 and induce apoptosis in RVFV infected liver cells. Virology. 2014;449:270–286. doi: 10.1016/j.virol.2013.11.023
- Quellec J, Pedarrieu A, Piro-Megy C, et al. Rift valley fever virus modulates apoptosis and immune response during infection of human astrocytes. Emerg Microbes Infect. 2023;12(1):2207672. doi: 10.1080/22221751.2023.2207672
- Michaely LM, Rissmann M, Armando F, et al. Rift valley fever virus non-structural protein S is associated with nuclear translocation of active caspase-3 and inclusion body formation. Viruses. 2022;14(11):14. doi: 10.3390/v14112487
- Qu B, Qi X, Wu X, et al. Suppression of the interferon and nf-κB responses by severe fever with thrombocytopenia syndrome virus. J Virol. 2012;86(16):8388–8401. doi: 10.1128/JVI.00612-12
- Song P, Zheng N, Liu Y, et al. Deficient humoral responses and disrupted B-cell immunity are associated with fatal SFTSV infection. Nat Commun. 2018;9(1):3328. doi: 10.1038/s41467-018-05746-9
- Sun Q, Jin C, Zhu L, et al. Host responses and regulation by NFκB signaling in the liver and liver epithelial cells infected with a novel tick-borne bunyavirus. Sci Rep. 2015;5(1):11816. doi: 10.1038/srep11816
- Xu S, Jiang N, Nawaz W, et al. Infection of humanized mice with a novel phlebovirus presented pathogenic features of severe fever with thrombocytopenia syndrome. PLOS Pathog. 2021;17(5):e1009587. doi: 10.1371/journal.ppat.1009587
- Liu S, Liu H, Zhang K, et al. Proteasome inhibitor PS-341 effectively blocks infection by the severe fever with thrombocytopenia syndrome virus. Virol Sin. 2019;34(5):572–582. doi: 10.1007/s12250-019-00162-9
- Lozach PY, Kuhbacher A, Meier R, et al. DC-SIGN as a receptor for phleboviruses. Cell Host & Microbe. 2011;10(1):75–88. doi: 10.1016/j.chom.2011.06.007
- Wonderlich ER, Caroline AL, McMillen CM, et al. Peripheral blood biomarkers of disease outcome in a monkey Model of rift valley fever encephalitis. J Virol. 2018;92(3). doi: 10.1128/JVI.01662-17
- Zhang W, Li M, Xiong S, et al. Decreased myeloid dendritic cells indicate a poor prognosis in patients with severe fever with thrombocytopenia syndrome. Int J Infect Dis. 2017;54:113–120. doi: 10.1016/j.ijid.2016.11.418
- Song P, Zheng N, Zhang L, et al. Downregulation of interferon-β and inhibition of TLR3 expression are associated with fatal outcome of severe fever with thrombocytopenia syndrome. Sci Rep. 2017;7(1):6532. doi: 10.1038/s41598-017-06921-6
- Harmon JR, Spengler JR, Coleman-McCray JD, et al. CD4 T cells, CD8 T cells, and monocytes coordinate to prevent rift valley fever virus encephalitis. J Virol. 2018;92(24). doi: 10.1128/JVI.01270-18
- Hum NR, Bourguet FA, Sebastian A, et al. MAVS mediates a protective immune response in the brain to Rift Valley fever virus. PLOS Pathog. 2022;18(5):e1010231. doi: 10.1371/journal.ppat.1010231
- Lu Q-B, Cui N, Hu J-G, et al. Characterization of immunological responses in patients with severe fever with thrombocytopenia syndrome: a cohort study in China. Vaccine. 2015;33(10):1250–1255. doi: 10.1016/j.vaccine.2015.01.051
- Li M, Xiong Y, Li M, et al. Depletion but activation of CD56dimCD16+ NK cells in acute infection with severe fever with thrombocytopenia syndrome virus. Virol Sin. 2020;35(5):588–598. doi: 10.1007/s12250-020-00224-3
- Du J, Li XK, Peng XF, et al. Expansion of granulocytic myeloid‐derived suppressor cells in patients with severe fever with thrombocytopenia syndrome. J Med Virol. 2022;94(9):4329–4337. doi: 10.1002/jmv.27854
- Huang M, Wang T, Huang Y, et al. The clinical and immunological characteristics in fatal severe fever with thrombocytopenia syndrome virus (SFTSV) infection. Clin Immunol. 2023;248:248. doi: 10.1016/j.clim.2023.109262
- Sun L, Hu Y, Niyonsaba A, et al. Detection and evaluation of immunofunction of patients with severe fever with thrombocytopenia syndrome. Clin Exp Med. 2013;14(4):389–395. doi: 10.1007/s10238-013-0259-0
- Dodd KA, Ak M, Jones ME, et al. Rift valley fever virus clearance and protection from neurologic disease are dependent on CD4+ T cell and virus-specific antibody responses. J Virol. 2013;87(11):6161–6171. doi: 10.1128/JVI.00337-13
- Park A, Park SJ, Jung KL, et al. Molecular signatures of inflammatory profile and B-Cell function in patients with severe fever with thrombocytopenia syndrome. MBio. 2021;12(1):12. doi: 10.1128/mBio.02583-20
- Zhang J, Yan X, Li Y, et al. Reactive plasmacytosis mimicking multiple myeloma associated with SFTS virus infection: a report of two cases and literature review. BMC Infect Dis. 2018;18(1):18. doi: 10.1186/s12879-018-3431-z
- Takahashi T, Suzuki T, Hiroshige S, et al. Transient appearance of plasmablasts in the peripheral blood of Japanese patients with severe fever with thrombocytopenia syndrome. J Infect Dis. 2019;220(1):23–27. doi: 10.1093/infdis/jiz054
- Song P, Zheng N, Liu Y, et al. Deficient humoral responses and disrupted B-cell immunity are associated with fatal SFTSV infection. Nat Commun. 2018;9(1). doi: 10.1038/s41467-018-05746-9
- Li M-M, Zhang W-J, Weng X-F, et al. CD4 T cell loss and Th2 and Th17 bias are associated with the severity of severe fever with thrombocytopenia syndrome (SFTS). Clin Immunol. 2018;195:8–17. doi: 10.1016/j.clim.2018.07.009
- Yi X, Li W, Li H, et al. Circulating regulatory T cells in patients with severe fever with thrombocytopenia syndrome. Infect Dis (Auckl). 2015;47(5):294–301. doi: 10.3109/00365548.2014.987812
- Li XK, Lu QB, Chen WW, et al. Arginine deficiency is involved in thrombocytopenia and immunosuppression in severe fever with thrombocytopenia syndrome. Sci Transl Med. 2018;10(459):10. doi: 10.1126/scitranslmed.aat4162
- Xu H, Wei Y, Ma H, et al. Alterations of gut microbiome in the patients with severe fever with thrombocytopenia syndrome. Front Microbiol. 2018;9:2315. doi: 10.3389/fmicb.2018.02315
- Xie J, Li H, Zhang X, et al. Akkermansia muciniphila protects mice against an emerging tick-borne viral pathogen. Nat Microbiol. 2023;8(1):91–106. doi: 10.1038/s41564-022-01279-6
- Zhang L, Wang D, Shi P, et al. A naturally isolated symbiotic bacterium suppresses flavivirus transmission by Aedes mosquitoes. Science. 2024;384(6693):eadn9524. doi: 10.1126/science.adn9524
- Sun Y, Chen C, Zeng C, et al. Severe fever with thrombocytopenia syndrome virus infection shapes gut microbiome of the tick vector haemaphysalis longicornis. Parasit Vectors. 2024;17(1):107. doi: 10.1186/s13071-024-06204-w
- Tchouassi DP, Muturi EJ, Arum SO, et al. Host species and site of collection shape the microbiota of Rift Valley fever vectors in Kenya. PLOS Negl Trop Dis. 2019;13(6):e0007361. doi: 10.1371/journal.pntd.0007361
- Gowen BB, Hickerson BT. Hemorrhagic fever of bunyavirus etiology: disease models and progress towards new therapies. J Microbiol. 2017;55(3):183–195. doi: 10.1007/s12275-017-7029-8
- Gai ZT, Zhang Y, Liang MF, et al. Clinical progress and risk factors for death in severe fever with thrombocytopenia syndrome patients. J Infect Dis. 2012;206(7):1095–1102. doi: 10.1093/infdis/jis472
- Li YH, Wang XH, Huang WW, et al. RETRACTED: severe fever with thrombocytopenia syndrome virus induces platelet activation and apoptosis via a reactive oxygen species-dependent pathway. Redox Biol. 2023;65:102837. doi: 10.1016/j.redox.2023.102837
- Fang L, Yu S, Tian X, et al. Severe fever with thrombocytopenia syndrome virus replicates in platelets and enhances platelet activation. J Thromb Haemost. 2023;21(5):1336–1351. doi: 10.1016/j.jtha.2023.02.006