
ABSTRACT
Glioblastoma is an aggressive and incurable form of brain cancer. Both mutation analysis in human glioblastoma and mouse modelling studies have shown that aberrant activation of the PI 3-kinase pathway is a central driver of glioblastoma malignancy. The small GTPase Rac is activated downstream of this pathway, mediating a subset of the effects of aberrant PI 3-kinase pathway activation. Here I discuss the current state of our knowledge on Rac activation mechanisms in glioblastoma. Current knowledge on roles for specific PI 3-kinase pathway responsive Rac guanine nucleotide exchange factors in glioblastoma is reviewed. Rac is best known for its role in promoting cell motility and invasion, but there is also evidence for roles in multiple other cellular processes with cancer relevance, including proliferation, differentiation, apoptosis, DNA damage responses, metabolism, angiogenesis and immunosuppression. I review what is known about the role of Rac in these processes in glioblastoma. Finally, I assess possible strategies to inhibit this pathway in glioblastoma through either direct inhibition of Rac or inhibition of upstream activators or downstream mediators of Rac signalling.
KEYWORDS:
Overview of glioblastoma
Glioblastoma (grade IV astrocytoma) is the most common form of adult brain tumour, with an age-adjusted incidence of 0.6–3.7 per 100,000 persons [Citation1]. It is one of the most aggressive cancers, with median survival time after diagnosis of only 12–14 months and a five-year survival rate of less than 5% [Citation1] (https://www150.statcan.gc.ca/n1/pub/82-226-x/82-226-x2012001-eng.htm). Clinically, glioblastoma can be divided into two types: primary glioblastoma and secondary glioblastoma. Primary glioblastoma presents without any previous history of lower grade brain cancer. Secondary glioblastoma occurs in patients with a previous history of lower grade astrocytoma and is much less common. Primary and secondary glioblastoma have distinct genetic changes and from a molecular perspective can be thought of as different diseases [Citation2]. This review focuses on primary glioblastoma. With the exception of exposure to high levels of radiation in childhood, which accounts for a very small number of cases, there are no known environmental factors that cause glioblastoma [Citation3]. For this reason, much current glioblastoma research focuses on improving treatment.
Glioblastoma pathology
Definitive diagnosis of glioblastoma comes from analysis of surgical tissue by a neuropathologist [Citation4]. Diagnostic features of glioblastoma include hypercellularity with mitotic figures, nuclear atypia, regions of necrosis and microvasculature proliferation. In additional to histological features, molecular analyses such as IDH mutation status may be used to refine the diagnosis [Citation2]. Diffuse growth is also a defining feature of glioblastoma. Tumour borders are invariably diffuse and in rare cases, glioblastoma can present without any central mass, a growth pattern known as gliomatosis cerebri [Citation5]. Glioblastoma rarely metastasizes outside the CNS, but is highly invasive within this compartment, migrating as single cells along white matter tracts, the outside walls of blood vessels and the subpial space. Recent work has shown that, in at least a subset of glioblastomas, these single cells are often connected by long projections known as microtubes [Citation6]. These allow direct communication between glioblastoma cells via gap junctions and appear to have a role in promoting invasion.
Glioblastoma therapy
Standard treatment for both primary and secondary glioblastoma typically includes surgery followed by radiation and chemotherapy with temozolomide, an alkylating agent developed for efficient transfer across the blood/brain barrier [Citation7]. Radiation and chemotherapy are not curative. Multiple mechanisms for resistance have been identified. A subset of glioblastoma cells with neural stem cell-like characteristics contributes to resistance to both radiation and temozolomide [Citation8,Citation9], at least in part due to increased activity of DNA damage response pathways in these cells [Citation9]. Microtube connections have also been proposed to contribute to glioblastoma resistance to radiation and chemotherapy [Citation6,Citation10]. Glioblastoma is also a very heterogeneous cancer. This was evident early on from histological, immunohistochemical and fluorescence in situ hybridization studies, and has been studied in detail using single-cell sequencing technologies [Citation11]. Both phenotypic heterogeneity and genetic heterogeneity probably contribute to resistance to standard therapy. Molecular heterogeneity also very likely explains the failure of tyrosine kinase inhibitors in this disease, as single cell analysis showed the use of different tyrosine kinase receptors by different cancer cells in the same patient [Citation11]. Glioblastoma has also to date been resistant to the immune checkpoint inhibitors that have been successful in other cancer types [Citation12]; potential mechanisms for this are described in detail later in this review.
Mutational profile of glioblastoma
Early karyotypic analyses of glioblastoma showed characteristic chromosomal gains and losses and more recently comprehensive analyses of large numbers of glioblastoma cases using microarray analyses for copy number have given a more comprehensive picture of this [Citation13,Citation14]. The gain of chromosome 7 is a near-universal event in primary glioblastoma, as is the loss of one copy of chromosome 10 [Citation15]. These are probably early events in primary glioblastoma genesis [Citation15]. Loss of the short arm of chromosome 9 is also very common [Citation13]. On chromosome 7, EGFR is very commonly amplified [Citation13,Citation14]. A second tyrosine kinase receptor, MET, is also on chromosome 7 and is amplified in a small percentage of cases [Citation14]. Other less common amplifications (not on chromosome 7) include MDM4 (7%), PDGFRA (10%) and CDK4 (14%) [Citation14]. PTEN and CDKN2A are the most commonly mutated tumour suppressors in glioblastoma. Ninety per cent of primary glioblastomas have lost at least one copy of PTEN (usually due to the loss of an entire copy of chromosome 10) and 40% have loss or inactivation of both alleles [Citation16]. Through a combination of large and focal deletions on chromosome 9, loss of one allele of CDKN2A occurs in 80% of cases and loss of both alleles in 60% of cases. Less common tumour suppressor mutations are found in TP53 (28%), RB1(8%) and NF1(10%). Common mutations that activate oncogenes in glioblastoma are found for EGFR (40%) and PI 3-kinase family genes (25% total, predominately in PIK3CA and PIK3R1) [Citation14]. Activating mutations in RAS genes and BRAF are rare (1–2%). TERT promoter mutations, which increase telomerase expression and activity, are found in over 80% of primary glioblastomas [Citation14,Citation17].
Integration of the mutational profile for glioblastoma shows a key role for aberrant PI 3-kinase pathway activation. While receptor tyrosine kinases activate multiple signalling pathways in addition to the PI 3-kinase pathway, the loss of at least one copy of PTEN in 90% of primary glioblastomas, together with further activation of this pathway via inactivation of the second copy of PTEN (40% of cases) and PI 3-kinase family genes (25% of cases and mutually exclusive with PTEN deletions/mutations) indicates a key role for this pathway. The role of Akt signalling as a downstream mediator in the PI 3-kinase pathway has received a great deal of attention. However, it has long been known that additional, Akt-independent pathways, are also important in mediating the oncogenic effects of PI 3-kinase pathway signalling [Citation18,Citation19]. One such pathway is signalling through the small GTPase Rac. The overall contribution of PI 3-kinase pathway activation of Rac to glioblastoma malignancy is not fully understood; this review focuses on summarizing what is currently known.
Rac family members and their biochemistry
Rac proteins are classified as a subfamily of the Rho family of small G proteins. The Rac subfamily has four members, Rac1, Rac2, Rac3 and RhoG; these are encoded by genes on chromosome 7, 22, 17 and 11, respectively. They are all GTPases that switch between an active GTP-bound state and an inactive GDP-bound state. Activation occurs when a guanine nucleotide exchange factor protein (GEF) displaces bound GDP from Rac, allowing GTP to bind in its place. Rac is inactivated by GTPase activating proteins. A third layer of control over Rac activation is imparted by guanine nucleotide inhibitor proteins (GDIs), which block Rac activation by inhibition of GDP dissociation and cytoplasmic sequestration [Citation20].
An alternate form of Rac1, known as Rac1b, is also expressed through alternate splicing. This has an insertion of an additional 19 amino acids due to a fifty-seven-nucleotide insertion between exons 3 and 4 in the mRNA [Citation21–23]. Rac1b has enhanced GTP/GDP exchange and impaired GTPase activity and is therefore constitutively active in the absence of Rac GEFs [Citation24]. Rac1b is preferentially expressed in several cancer types, has altered engagement of downstream signalling pathways and has transforming activity in NIH3T3 cells [Citation21,Citation23,Citation25,Citation26]. Alternate splicing to generate Rac1b is mediated by activation of the splicing factor SRSF1 by SRPK1 kinase [Citation27]. This pathway is activated downstream of the EGF receptor in HeLa cells [Citation28].
Rac family members in glioblastoma
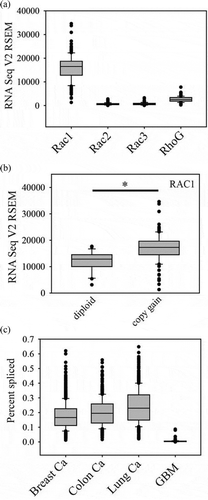
RNA-seq data from the TCGA database shows that Rac1 mRNA is expressed at higher levels than Rac2, Rac3 or RhoG in glioblastoma ()) [Citation14,Citation29–31]. The RAC1 gene is on the short arm of chromosome 7 (7p22.1) and so there is RAC1 copy gain in most glioblastomas. It is not usually part of the EGFR (7p11.2) amplicon and is only amplified in 1% of glioblastomas. However, the copy gain does correspond to increased mRNA expression ()) and this may, therefore, contribute to increased Rac1 activity in glioblastoma. Given the previously mentioned concept that chromosome 7 gain and chromosome 10 loss are early events in gliomagenesis [Citation15], this suggests that aberrant PI 3-kinase-mediated Rac1 activation, through the loss of one copy of PTEN and RAC1 copy gain, may be an early and very common event in primary glioblastoma. The activating P29S RAC1 mutations that have been detected in 5–10% of melanomas [Citation32,Citation33] do not appear to be present in glioblastoma. Although present in many other cancer types, Rac1b also does not appear to be expressed in glioblastoma, based on analysis of the TCGA database with TCGASpliceSeq [Citation34] ()).
Figure 1. Rac mRNA expression in glioblastoma. (a). mRNA expression levels for Rac1, Rac2, Rac3 and RhoG in the TCGA 2013 glioblastoma database were analyzed using cBioPortal. RSEM normalized data are shown. (b). Rac1 mRNA levels in cases with copy gain (n = 110, 80% of cases) compared to cases without copy gain (n = 27). (c). Frequency of Rac1b splicing in TCGA database samples was analyzed using TCGA SpliceSeq in breast invasive carcinoma, colon adenocarcinoma, lung adenocarcinoma and glioblastoma (GBM). Graphs were generated using SigmaPlot software. Box plots show the median, 10th, 25th, 75th and 90th percentiles. * indicates p < 0.05 by the Mann-Whitney Rank Sum test
Rac GEFs in glioblastoma
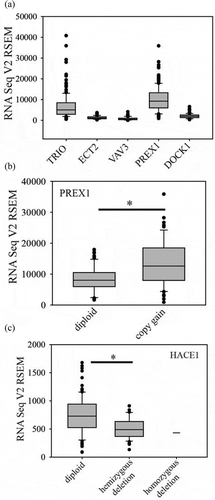
About twenty GEFs able to activate Rac1 have been identified [Citation35]. In addition to activating Rac, Rac GEFs have additional roles as scaffolds that guide Rac signalling along specific downstream pathways [Citation35]. For example, Marei et al. have shown that in the same context, different Rac GEFS have opposing effects on cell motility [Citation36]; in NIH 3T3 cells, TIAM1 expression inhibits cell migration, while PREX1 promotes cell migration, in part by forming a ternary complex with Rac1 and the actin remodelling protein flightless homolog II [Citation36]. This emphasizes that different Rac GEFs are not necessarily redundant and the types of Rac GEFs expressed may dictate Rac signalling outcomes. Rac GEFs that have been studied in glioblastoma include Trio, Ect2 and Vav3 [Citation37], PREX1 [Citation38] and DOCK1 [Citation39,Citation40]. The mRNA expression levels for these Rac GEFS in glioblastoma are shown in ). PREX1 mRNA expression is highest. Trio is also highly expressed, while Ect2, Vav3 and DOCK1 show only very low levels of expression. PREX1 protein is also expressed at high levels in glioblastoma; immunohistochemistry analysis of a tissue microarray showed that it is overexpressed relative to adjacent normal tissue in 90% of glioblastoma cases [Citation38]. The reasons for the high expression of PREX1 in glioblastoma are not fully known. Copy gain in glioblastoma may contribute to this. The PREX1 gene is at 20q13.13. Gain of chromosome 20 is seen in about 40% of glioblastomas and this copy gain is associated with an increase in PREX1 mRNA levels ()). As PREX1 protein level is increased in over 90% of glioblastomas [Citation38], additional factors are probably contributing to increased PREX1 levels. The histone deacetylase inhibitor tricostatin A induces PREX1 expression in non-metastatic prostate cancer cells, pointing to a role for promoter acetylation in the control of PREX1 expression in these cells [Citation41]. A similar effect was seen in patient-derived glioblastoma cells with lower PREX1 expression [Citation38]. PREX1 expression is positively regulated by ERK/MAPK signalling in BRAF and NRAS-mutant melanoma [Citation42]. It is not known whether this also affects PREX1 expression in glioblastoma. The transcription factor ZEB1 promotes PREX1 expression in glioblastoma cells [Citation43]. Singh et al. have linked the high expression of ZEB1 (along with SOX2 and OLIG1) to common glioblastoma oncogenic mutations [Citation44]. Thus, oncogenic activation of ZEB1 expression may be a common mechanism for increasing PREX1 levels in glioblastoma, although further studies are needed to confirm this.
Figure 2. Rac GEF and HACE1 mRNA expression in glioblastoma. (a). Expression of Rac GEFs in glioblastoma. (b). PREX1 mRNA levels in cases with copy gain (n = 52, 37% of cases) compared to cases without copy gain (n = 87). C. HACE1 mRNA levels are decreased in cases with copy loss (n = 41, 30% of cases) compared to cases without copy loss (n = 96). * indicates p < 0.05 by the Mann-Whitney Rank Sum test
PREX1 is directly activated by binding of either PIP3 or G protein βγ subunits, with the binding of both synergistically and maximally promoting activation [Citation45,Citation46]. This, along with the high expression of PREX1, suggest that it is probably a predominant Rac GEF linking aberrant PI 3-kinase pathway activation to Rac1 activation in glioblastoma. The Rac GEF DOCK1 (dedicator of cytokinesis; Dock180) has also activates Rac1 in glioblastoma. DOCK1 functions as a bipartite GEF with a second protein, ELMO [Citation47]. DOCK1/ELMO were first shown to have a role in glioblastoma Rac activation by Jarzynka et al. [Citation39]. Further studies showed that PDGF and EGF receptors in glioblastoma cells activate DOCK1 via Src kinase-mediated phosphorylation [Citation40,Citation48]. DOCK1 also binds PIP3 via its DHR-1 domain [Citation49]. This has an important role in recruiting DOCK1 to sites where it can be further activated [Citation49–51]. DOCK1 is, therefore, a second GEF linking aberrant PI 3-kinase signalling to Rac activation in glioblastoma, although the low mRNA expression levels suggest that this may be a minor pathway ()). Although the Rac GEF Trio is abundantly expressed at the mRNA level in glioblastoma ()), it does not interact with PIP3 but rather is activated by Gαq protein-coupled receptors and thus does not provide a direct link between Rac and the PI 3-kinase pathway [Citation52].
Post-translational modifications of Rac1
Ubiquitination has emerged as an important mechanism for controlling Rac1 protein levels and activity. Three E3 ligases that mediate Rac1 ubiquitination have been identified: HACE1 [Citation53], IAP family members [Citation54] and SCF(FBXL19) [Citation55]. With all E3 ligases, ubiquitination leads to loss of Rac1 activity as a consequence of proteasomal degradation. HACE1 selectively mediates ubiquitination of activated Rac1 [Citation53], while IAPs and SCF(FBXL19) are active against both active and inactive Rac1 [Citation55]. HACE1 is a tumour suppressor that is silenced by genetic or epigenetic mechanisms in multiple cancers [Citation56]. It is on chromosome 6 at the q16.3/q21 boundary. The long arm of chromosome 6 is frequently deleted in glioblastoma and as a result, 30% of glioblastomas are haploinsufficient for HACE1, which results in decreased mRNA expression ()). Mouse models did not identify an effect of HACE1 haploinsufficiency in tumour incidence; however recent studies have suggested that HACE1 haploinsufficiency, combined with further epigenetic silencing of HACE1, contributes to the pathogenesis of lymphomas [Citation57,Citation58]. Although this has not been investigated, it seems likely that a similar mechanism contributes to glioblastoma pathogenesis, given the low levels of HACE1 mRNA in glioblastomas with either one or two copies of the HACE1 gene ()). Although HACE1 negatively regulates activated Rac1, activated Rac1 also negatively regulates HACE1 via group I PAK kinase-mediated phosphorylation [Citation59]: mutually antagonistic signalling networks such as this have been shown to generate “on-off’ switches for cellular functions [Citation60]. Rac1 is also modified by sumoylation, which enhances the activation of Rac1 and promotes lamellipodia formation [Citation61]. The significance of this in glioblastoma or other cancers is not currently known.
Feedback inhibition of Rac
One well-studied function for Rac in normal tissues is in migration of neutrophils towards a chemokine gradient. This requires an adaptation mechanism to allow Rac to respond to both low and high chemokine concentrations. Graziano et al. have used optogenetics to identify a feedback inhibition mechanism for Rac in neutrophils; this involves activation by PREX1 and inactivation by the Rac GAP ARHGAP15 [Citation62]. In glioblastoma and other cancers, this mechanism would presumably be inactive in order to allow sustained Rac signalling in response to a (likely) invariant input signal. TCGA database inspection shows that ARHGAP15 expression is very low in glioblastoma, suggesting one simple mechanism for achieving this.
Summary of Rac activation mechanisms in glioblastoma
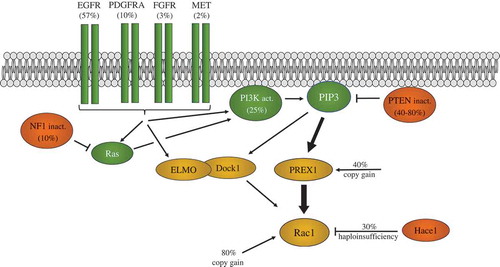
shows a summary of known common genetic changes in glioblastoma that can contribute to Rac1 activation. These include receptor tyrosine kinase amplifications (EGFR, PDGFR, FGFR, MET), inactivation of tumour suppressors (PTEN, NF1, HACE1), PI 3-kinase activating mutations and copy gains in the genes encoding the Rac GEF PREX1 and RAC1 itself. This suggests that aberrant Rac1 activation is probably a common element in most or all glioblastomas.
Figure 3. Glioblastoma genetic alterations promoting Rac1 activation. The Epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (a) (PDGFRA), the fibroblast growth factor receptor (FGFR) and the MET receptor (MET) genes are frequently amplified and/or mutated in glioblastoma. This can lead to activation of Rac1 via either PREX1 or ELMO/Dock1 Rac GEFs. PREX1 is directly activated by PIP3 binding, while ELMO/Dock1 requires PIP3 binding as part of its activation mechanism. High PREX1 mRNA and protein levels in glioblastoma suggest it may be the predominant Rac GEF used. PTEN inactivation by copy loss and/or mutation, as well as PI 3-kinase mutational activation, also promotes Rac1 activation via these Rac GEFs. NF1 mutation leads to Ras activation, which can also activate PI 3-kinase and Rac1. Copy gains of RAC1, and PREX1, as well as HACE1 copy loss, may also influence the extent of Rac1 activation. Frequencies of alterations in glioblastoma cases are shown in brackets
Rac contributions to the malignant behaviour of glioblastoma
Rac1 in motility and invasion
Rac has been widely studied with respect to its role in cell motility and cancer cell invasion. Cancer cells employ multiple mechanisms to invade neighbouring tissue, including lamellipodia and invadopodia [Citation63,Citation64]. The role of Rac1 in lamellipodia function has been studied in most detail [Citation63,Citation65]. Lamellipodia are large fan-like structures, the formation of which is principally driven by actin dynamics. Highly branched actin filaments are produced by the action of the Arp2/3 complex, which nucleates new branching actin filaments from existing actin filaments in an autocatalytic fashion where the product serves as the substrate for further formation of branched actin filaments [Citation63]. Arp2/3 activity is regulated by the multiprotein WAVE complex. GTP-bound Rac1 binds directly to two sites on the WAVE complex [Citation66–68], relieving the autoinhibited conformation of the latter and allowing it to activate Arp2/3. Rac1 also indirectly regulates the WAVE complex through its interaction with lamellipodin [Citation69]. Similar to its activation of WAVE, Rac1 also activates the protein Arpin; this inhibits Arp2/3 and actin polymerization, thereby regulating directional persistence of migration [Citation70]. On the basis of these multiple functions, Rac is considered a master regulator of lamellipodia. Invadopodia are a second mechanism used by cancer cells to invade tissue [Citation64]. Invadopodia are longer, thinner membrane protrusions than lamellipodia that are also found in normal cells, where they are called podosomes [Citation64]. Data from Rac1 and Rac2 knockout mice show a role for both in macrophage podosome formation and invasion [Citation71]; it seems likely that Rac family members also have a role in invadopodia-mediated tissue invasion of cancer cells. In addition to promoting motility, Rac1 can also increase extracellular matrix-degrading metalloprotease activity to promote the invasive behaviour of some cell types [Citation72].
Most studies on glioblastoma cell motility/invasion have focussed on the identification of roles for specific Rac GEFs. Trio, Ect2, Vav3, DOCK1/ELMO and PREX1 have all been proposed to have roles in glioblastoma invasion [Citation37–40]. Based on its expression, activation mechanism and known role in the invasive behaviour of other cancers, PREX1 appears to be the most likely candidate for PI 3-kinase pathway-driven motility/invasion in glioblastoma. It is highly expressed in patient-derived glioblastoma cells that retain their invasive behaviour after intracerebral injection into immunocompromised mice while being low or absent in standard glioblastoma cell lines that are non-invasive in vivo, and PREX1 knock-down in cell culture reduces motility and invasion in multiple primary patient-derived glioblastoma lines [Citation38].
Rac1 in proliferation and apoptosis
In addition to its role in cell motility and invasion, Rac1 has well-established roles in both proliferation and apoptosis. Rac1 has an essential role in the serum-induced G1/S progression of mouse fibroblasts [Citation73]. This is mediated by increased cyclin D1 transcription as a consequence of Rac1 activation of NFκB [Citation74,Citation75]. Rac1B, the constitutively active splice variant of Rac1, promotes cyclin D1 accumulation and G1/S progression in mouse fibroblasts via an NFκB-dependent mechanism [Citation76]. Activation of NFκB downstream of Rac1B was suggested to occur via NADPH oxidase [Citation76,Citation77]. The same study also showed that Rac1b (and also normal Rac1) was able to suppress apoptosis in mouse fibroblasts, consistent with other studies [Citation78,Citation79]. Senger et al. [Citation80] looked in detail at the role of Rac1 in glioblastoma cell apoptosis. They found that adenovirus-mediated expression of dominant negative Rac1 induced apoptosis in multiple glioblastoma cells lines and short-term patient glioblastoma cell cultures (grown in 10% fetal bovine serum), independent of the TP53, CDKN2A, MDM2 and EGFR mutation status of the cells. This did not occur in normal human astrocytes. Dominant-negative Rac1 induction of apoptosis was repressed by expression of a constitutively active version of MAP3K1 (MEKK1), an activator of Jnk and NFκB [Citation80].
Rac1 in differentiation
Several studies have linked Rac1 activity to the maintenance of a stem cell-like state in tissues and cancer. In normal tissues, the deletion of Rac1 in mouse epidermis caused stem cells to undergo terminal differentiation [Citation81]. This was due to the negative regulation of cMyc by PAK2-mediated phosphorylation [Citation81]. In a mouse model of sebaceous skin cancer, activation of Rac1 did not affect cancer incidence but did result in the formation of tumours that, along with being more invasive and proliferative, were less differentiated [Citation82]. Yoon et al. have reported that Rac1 knock-down by RNA interference reduces both the neural stem cell-like characteristics and malignant properties of neurospheres derived from U87MG and U373 [Citation83], suggesting that Rac1 may also promote or maintain a neural stem cell-like phenotype in glioblastoma. More recently, Almiron Bonnin et al. showed that treatment of glioma stem-like cells with the Rac inhibitor EHT 1864 reduced their self-renewal [Citation84].
Rac1 in DNA damage responses
As mentioned earlier, radiation with concomitant administration of the alkylating agent temozolomide is the standard of care for glioblastoma. This improves survival but is not curative. Bao et al. [Citation9]. have shown that glioblastoma cells with neural stem cell-like characteristics exhibit intrinsic resistance to DNA damage, showing both an enhanced activation of the DNA damage response and a greater capacity for DNA repair. This publication did not describe a mechanism for this. Pak1, activated by Rac, has been shown to promote resistance to DNA damage by several mechanisms [Citation85]. Pak1 phosphorylates CRAF, which is then able to recruit and activate the cell cycle checkpoint/DNA damage response kinase Chk2 [Citation86]. In addition, PAK1 phosphorylates the protein MORC2 to promote DNA repair via γH2AX induction [Citation87]. Whether these pathways operate in glioblastoma has not been determined.
Rac1 in metabolism
Enhanced glycolysis has long been recognized as a common feature of cancer cells and activation of receptor tyrosine kinases such as the insulin receptor promote glycolysis via activation of the PI 3-kinase pathway. Uptake and phosphorylation of glucose are promoted by Akt activation downstream of this pathway [Citation88]. However, recent evidence shows that PI 3-kinase pathway-mediated enhancement of glycolysis also requires activation of aldolase. This occurs by a pathway that is independent of Akt but instead requires Rac [Citation89]. Actin remodelling induced by Rac activation results in the release of aldolase A from actin filaments, which otherwise bind aldolase and keep it in an inactive state [Citation89]. Specific roles for Rac GEFs in this process have not been characterized to date and the importance of this pathway specifically in glioblastoma metabolism has not been assessed. Studies in orthotopic mouse models and humans show that glioblastomas are very active in glycolysis and use the pyruvate produced during glycolysis to generate intermediates for macromolecular biosynthesis [Citation90,Citation91]. Thus, Rac, via aldolase activation, may have an important role in facilitating glioblastoma growth by supporting the production of the lipids and amino acids required for cell proliferation [Citation92].
Rac1 activation of NADPH oxidase produces superoxide, which in turn generates other reactive oxygen species [Citation93]. High Rac1 activity can therefore potentially lead to susceptibility to reactive oxygen species-induced cell death. One of the key molecules cells use to protect cells from this fate is glutathione, which neutralizes reactive oxygen species. As glutathione is synthesized from TCA cycle intermediates, this can lead to ‘addiction’ of cancer cells to high levels of glycolysis, such that restricting this metabolic pathway leads to ROS-mediated cell death [Citation94]. Thus, Rac activation may provide avenues to selectively kill glioblastoma cells through inhibition of metabolic enzymes.
Rac1 in angiogenesis
Glioblastoma is one of the most highly vascularized tumours known [Citation95]. There is evidence for a direct role for Rac1 in multiple aspects of angiogenesis (reviewed in [Citation96]). Vader et al. showed that knock-down of Rac1 in human endothelial cells inhibited their migration, invasion, proliferation and tube formation both in cell culture and in vivo in a Matrigel plug assay [Citation97]. However, at least in mice, this role for Rac1 can be redundant with a Rac1-independent signalling pathway operating through α5β3 integrin [Citation98]. Based on inhibitor data, a recent study supports a role for Rac family members in the secretion of VEGF by glioblastoma cells [Citation84], suggesting that Rac functions in both glioblastoma cells and tumour endothelial cells may be required for glioblastoma angiogenesis.
Rac1 in immunosuppression
Glioblastoma is described as an immunologically ‘cold’ cancer [Citation99]. Glioblastoma cells secrete cytokines that attract microglia, macrophages and myeloid-derived suppressor cells; in some cases, myeloid cells can make up half of the cells present in a glioblastoma tumour [Citation100]. The tumour-associated macrophages adopt an immunosuppressive phenotype under the influence of glioblastoma cells. Glioblastoma cytokine secretion also recruits Tregs, which further contribute to the immunologically “cold’ nature of glioblastoma [Citation99]. T cells with the potential to elicit anticancer activity are inactivated by multiple mechanisms in glioblastoma [Citation101], including a recently described mechanism that promotes their sequestration in the bone marrow [Citation102]. With respect to signalling pathways, STAT3 has a central role in promoting the immunosuppressive environment in glioblastoma and other cancers [Citation103]. In cancer cells, STAT3 is activated by multiple inputs including EGFR signalling. This leads to the expression of cytokines such as VEGF and IL-10 that in turn activate STAT3 in immune cells. This promotes immunosuppression by multiple mechanisms including the suppression of dendritic cell maturation [Citation104], the maintenance and activity of myeloid-derived suppressor cell populations [Citation105], the induction of an immunosuppressive M2-like phenotype in macrophages [Citation106], suppression of NK cell activity [Citation107] and the expansion of tumor-infiltrating Treg populations [Citation107]. Rac1 is known to have a role in STAT3 activation [Citation108]. Both direct and indirect (via IL6 induction) mechanisms have been proposed [Citation109–111]. This raises the possibility that signalling through Rac1 has a central role in establishing the immunosuppressive environment in glioblastoma, although direct evidence for this is lacking.
Targeting Rac signalling in glioblastoma
The literature described above suggests that Rac1 may have a central role in many aspects of glioblastoma malignant behaviour. Inhibition of this pathway may be beneficial directly or may sensitize glioblastoma to other therapies that are ineffective as single agents. Several recent reviews have covered the general field of Rac inhibitors [Citation112,Citation113]. Studies with relevance to glioblastoma are described in the following sections.
Direct inhibition of Rac1 GTP binding
EHT 1864 was first characterized as a Rac1 pathway inhibitor that reduced the processing of amyloid precursor proteins [Citation114]. Further characterization suggested that EHT 1864 was able to bind directly to various Rac family members with nanomolar affinities, inhibiting nucleotide association and maintaining Rac in an inactive state [Citation115]. However, a binding site on Rac for EHT 1864 has never been determined, which would be a critical step in validating a direct interaction, and so the mechanism of action for EHT 1864 is not clearly established. EHT 1864 appears to block Rac, but not Cdc42 or RhoA, engagement with downstream effectors [Citation114,Citation115]. Using focus-forming assays as a readout, EHT 1864 blocked Rac1-induced NIH 3T3 fibroblast transformation, as well as Ras transformation, which is Rac-dependent in NIH 3T3 cells [Citation115–117]. Almiron Bonnin et al. have evaluated EHT 1864 in a mouse model of high-grade glioma [Citation84]. S100β-v-erbB/p53−/- mice were used in this study, which develops a cancer similar to human high-grade oligodendroglioma [Citation118]. In this model, EHT 1864 treatment was found to reduce VEGF secretion by glioma cells both in cell culture and in vivo. When administered intraperitoneally, it also significantly reduced the tumour growth of glioma cells implanted subcutaneously in mice. To date, there are no reports evaluating EHT 1864 in models of primary glioblastoma. Other compounds that directly target Rac are being explored but are generally at very early stages of development (reviewed in Maldonado and Dharmawardhane [Citation112]).
Inhibition of Rac1/GEF interactions
Using a virtual screen, NSC23766 was identified as a compound that bound Rac1 at a site that was involved its interaction with the Dbl family GEFs Tiam1 and Trio [Citation119]. Karpel-Massler et al. found that NSC23766 worked either synergistically or additively with erlotinib and other EGF receptor inhibitors to inhibit the growth of a panel of glioblastoma cell lines [Citation120]. This combination also appeared to give a small increase in survival when administered intraperitoneally in an intracranial xenograft mouse model using glioblastoma cells isolated under serum-free conditions from a patient. However, NSC23766 has low potency, with an IC50 in the 50–100 uM range, and exhibited off-target effects, suggesting that it is unlikely to be a clinically useful drug [Citation112]. EHop-016 was developed as a more potent derivative of NSC23766, with an IC50 of 1 uM for Rac activation [Citation121]. This showed activity in a mouse xenograft model of metastatic breast cancer [Citation122], but there are no reports of it being tested in glioblastoma models. A derivative of EHop-016, MBQ-167 has 100 nM for inhibition of both Rac1 and Cdc42. Again, this is active in a metastatic mouse xenograft model of breast cancer [Citation123] but does no appear to have been tested in glioblastoma models. 1A-116 is another inhibitor of Rac/GEF interactions that was developed from a different pharmacophore from the above compounds [Citation124]. This has been shown to block PREX1/Rac1 interactions with an IC50 of 4 uM [Citation124]. 1A-116 has been shown to inhibit proliferation and promote apoptosis in two glioma cell lines [Citation125] but has not been evaluated in animal models of glioblastoma.
GEF inhibition
While the inhibitors described above bind to Rac1 to interfere with Rac GEF interactions, it may also be possible to target Rac GEFS, either directly or indirectly, through their upstream activation signals [Citation126]. C21 was identified as an inhibitor of DOCK5-mediated Rac activation [Citation127]. Although the precise binding site has not been mapped, studies suggest that C21 binds to a transient conformation of DOCK5 to prevent its activation of complexed Rac [Citation128]. This may illustrate the principle of direct Rac GEF inhibition, which could be further explored with other Rac GEFs implicated in glioblastoma pathogenesis.
Indirect inhibition of Rac GEF function can be achieved by inhibition of activating inputs, although this would generally inactivate multiple other signalling pathways as well. PREX1 requires two inputs for full activation: binding of PIP3 generated by the PI 3-kinase pathway, and binding of G protein βγ subunits from G protein-coupled receptor activation [Citation129]. Trials of PI 3-kinase inhibitors in glioblastoma have been reviewed recently [Citation130]. To date they have not shown promise due to a number of factors including on-target toxicities and poor CNS presentation; these may be addressable through the use of isoform-selective inhibitors with improved ability to cross the blood/brain barrier. The fact that PREX1 requires a second input from G protein-coupled receptors for activation may provide a second avenue to inhibit this GEF, with some evidence suggesting that dopamine receptors may contribute to PREX1 activation in glioblastoma [Citation38].
PAK inhibition
p21-activated kinases mediate many downstream effects of Rac1 activation and there has been significant interest in developing selective inhibitors for these enzymes [Citation131]. The recent demonstration that group I PAK kinases repress the ubiquitination and degradation of Rac1 [Citation59] suggests an even greater potential for this strategy, as PAK inhibition could lead to degradation of Rac1 and, consequentially, inhibition of signalling through multiple downstream effectors. To date the compound FRAX 597 probably has the best combination of selectivity, potency and stability for Group I PAK kinase inhibition, with IC50s for purified PAK1, PAK2 and PAK3 in the 8–19 nm range and no inhibition of Group II PAKs at 10 uM [Citation132]. This compound has shown activity in preclinical models of neurofibromatosis Type 2-associated schwannomas [Citation132] and KRAS-driven squamous cell carcinoma [Citation133] but has not been evaluated for glioblastoma therapy. FRAX486 is a related compound with low nanomolar IC50s for Group I PAK kinases [Citation134]. Unlike FRAX597, FRAX486 has been shown to cross the blood-brain barrier, reaching PAK kinase inhibitory levels in the brain within 1 h of a subcutaneous injection in mice [Citation134]. This compound shows therapeutic activity in a mouse model of Fragile X syndrome [Citation134] and also in patient-derived xenograft models of childhood acute lymphoblastic leukaemia [Citation135], but again has not been evaluated in glioblastoma animal models.
Summary
Current therapies for glioblastoma generally only provide modest improvements in survival. There are multiple challenges in the development of more effective therapies for this disease, including the well-documented heterogeneity of this cancer, its extensive invasion, its anatomical location in a delicate and essential structure protected by the blood-brain barrier, and its profound local and systemic immunosuppression. We now appear to have a detailed picture of the genetic changes in glioblastoma that can be used to inform new strategies. While different glioblastoma patients, and different glioblastoma cells within the same patient, have different genetic alterations, a common set of core pathways is activated in most glioblastomas and targeting non-redundant convergence points within these pathways may be an effective strategy. The common mutations and chromosomal changes in glioblastoma, combined with our knowledge of the signalling pathways involved, suggest that Rac1 activation may be one such non-redundant convergence point. This activation is likely to be aberrantly strong and sustained, and as such may have cancer-specific aspects that can be exploited therapeutically.
Although best known for its role in invasion, Rac1 potentially has a role in conferring many of the hallmarks of cancer, as outlined above. Rac1 inhibition might be effective either on its own or as a means to improve benefit from standard or novel therapies. A potential issue with targeting non-redundant convergence points in signalling pathways is the higher risk of unacceptable side effects. Possible issues to deal with this are better local delivery strategies or more selective strategies for targeting specific Rac1 activation or effector mechanisms. Local delivery strategies have been reviewed elsewhere [Citation136]. More selective targeting strategies hold promise. Key limitations here are that: (1) our understanding of the role of specific Rac1 GEFs and effectors in glioblastoma is still fairly poor; (2) there are relatively few compounds available to target these aspects of Rac1 signalling and even fewer that have good pharmacological properties. This involves targeting protein/protein interfaces, which is generally viewed as challenging. However, several reviewers [Citation126,Citation137]have made the point that the natural compound Brefeldin A potently inhibits the activation of the GTPase Arf by Arf GEFs, showing high selectivity for specific Arf and ArfGEF family members [Citation138]. Brefeldin A binds in the cleft formed by the complex of these two proteins, a mechanism referred to as interfacial inhibition [Citation137]. This suggests that the development of highly selective inhibitors may be possible with the careful design of screening strategies and the use of chemical libraries that explore diverse chemical space.
Acknowledgments
Thank you to the reviewers for their very helpful comments on this manuscript.
Disclosure statement
No potential conflict of interest was reported by the author.
Additional information
Funding
References
- Tamimi AF, Juweid M. Epidemiology and outcome of glioblastoma. In: De Vleeschouwer S, editor. Glioblastoma. Brisbane (AU): Codon Publications Copyright: The Authors; 2017. Available from: https://www-ncbi-nlm-nih-gov.proxy.bib.uottawa.ca/books/NBK470003/ doi: 10.15586/codon.glioblastoma.2017.ch8
- Appin CL, Brat DJ. Molecular pathways in gliomagenesis and their relevance to neuropathologic diagnosis. Adv Anat Pathol. 2015 Jan;22(1):50–58. . PubMed PMID: 25461780; eng.
- Hanif F, Muzaffar K, Perveen K, et al. Glioblastoma multiforme: a review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac J Cancer Prev. 2017 Jan 1;18(1):3–9. PubMed PMID: 28239999; PubMed Central PMCID: PMCPMC5563115. eng.
- Wick W, Osswald M, Wick A, et al. Treatment of glioblastoma in adults. Ther Adv Neurol Disord. 2018;11:1756286418790452. PubMed PMID: 30083233; PubMed Central PMCID: PMCPMC6071154. eng.
- Ranjan S, Warren KE. Gliomatosis cerebri: current understanding and controversies. Front Oncol. 2017;7:165. PubMed PMID: 28824876; PubMed Central PMCID: PMCPMC5545748. eng.
- Osswald M, Jung E, Sahm F, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015 Dec 03;528(7580):93–98. PubMed PMID: 26536111; eng.
- Stupp R, Mason WP, van Den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005 mar 10;352(10):987–996. 352/10/987 [pii].
- Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012 Aug 23;488(7412):522–526. nature11287 [pii].
- Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006 Dec 7;444(7120):756–760. nature05236 [pii].
- Weil S, Osswald M, Solecki G, et al. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. 2017 Apr 17 PubMed PMID: 28419303; eng. DOI:10.1093/neuonc/nox070.
- Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014 June 20;344(6190):1396–1401. science.1254257 [pii].
- Maxwell R, Jackson CM, Lim M. Clinical trials investigating immune checkpoint blockade in glioblastoma. Curr Treat Options Oncol. 2017 Aug;18(8):51. . PubMed PMID: 28785997; eng.
- Beroukhim R, Getz G, Nghiemphu L, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007 Dec 11;104(50):20007–20012. PubMed PMID: 18077431; PubMed Central PMCID: PMCPMC2148413. eng.
- Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013 Oct 10;155(2):462–477. S0092-8674(13)01208-7 [pii].
- Ozawa T, Riester M, Cheng YK, et al. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014 Aug 11;26(2):288–300. S1535-6108(14)00265-7 [pii].
- Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010 Jan 19;17(1):98–110. S1535-6108(09)00432-2 [pii].
- Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013 April 9;110(15):6021–6026. 1303607110 [pii].
- Lien EC, Dibble CC, Toker A. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol. 2017 Apr;45:62–71. PubMed PMID: 28343126; PubMed Central PMCID: PMCPMC5482768. eng.
- Fruman DA, Chiu H, Bd H, et al. The PI3K pathway in human disease. Cell. 2017 Aug 10;170(4):605–635. PubMed PMID: 28802037; eng.
- Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand‘: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011 Jul 22;12(8):493–504. . PubMed PMID: 21779026; PubMed Central PMCID: PMCPMC3260518. eng.
- Jordan P, Brazao R, Boavida MG, et al. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene. 1999 Nov 18;18(48):6835–6839. PubMed PMID: 10597294; eng.
- Matos P, Skaug J, Marques B, et al. Small GTPase Rac1: structure, localization, and expression of the human gene. Biochem Biophys Res Commun. 2000 Nov 2;277(3):741–751. PubMed PMID: 11062023; eng.
- Schnelzer A, Prechtel D, Knaus U, et al. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene. 2000 Jun 15;19(26):3013–3020. PubMed PMID: 10871853; eng.
- Fiegen D, Haeusler LC, Blumenstein L, et al. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J Biol Chem. 2004 Feb 6;279(6):4743–4749. PubMed PMID: 14625275; eng.
- Matos P, Collard JG, Jordan P. Tumor-related alternatively spliced Rac1b is not regulated by Rho-GDP dissociation inhibitors and exhibits selective downstream signaling. J Biol Chem. 2003 Dec 12;278(50):50442–50448. . PubMed PMID: 14506233; eng.
- Singh A, Karnoub AE, Palmby TR, et al. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene. 2004 Dec 16;23(58):9369–9380. PubMed PMID: 15516977; eng.
- Goncalves V, Henriques AF, Pereira JF, et al. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. Rna. 2014 Apr;20(4):474–482. PubMed PMID: 24550521; PubMed Central PMCID: PMCPMC3964909. eng.
- Wang F, Fu X, Chen P, et al. SPSB1-mediated HnRNP A1 ubiquitylation regulates alternative splicing and cell migration in EGF signaling. Cell Res. 2017 Apr;27(4):540–558. PubMed PMID: 28084329; PubMed Central PMCID: PMCPMC5385621. eng.
- Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012 May;2(5):401–404. 2/5/401 [pii].
- Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013 April 2;6(269):l1. scisignal.2004088 [pii].
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics. 2011 Aug 4;12:323.
- Alan JK, Lundquist EA. Mutationally activated Rho GTPases in cancer. Small GTPases. 2013 Jul -Sep;4(3):159–163. . PubMed PMID: 24088985; PubMed Central PMCID: PMCPMC3976972. eng.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012 Jul 20;150(2):251–263. PubMed PMID: 22817889; PubMed Central PMCID: PMCPMC3600117. eng.
- Ryan M, Wong WC, Brown R, et al. TCGASpliceSeq a compendium of alternative mRNA splicing in cancer. Nucleic Acids Res. 2016 Jan 4;44(D1):D1018–D1022. PubMed PMID: 26602693; PubMed Central PMCID: PMCPMC4702910. eng.
- Marei H, Malliri A. GEFs: dual regulation of Rac1 signaling. Small GTPases. 2017 Apr 03;8(2):90–99. . PubMed PMID: 27314616; PubMed Central PMCID: PMCPMC5464116. eng.
- Marei H, Carpy A, Woroniuk A, et al. Differential Rac1 signalling by guanine nucleotide exchange factors implicates FLII in regulating Rac1-driven cell migration. Nat Commun. 2016 Feb 18;7:10664. PubMed PMID: 26887924; PubMed Central PMCID: PMCPMC4759627. eng.
- Salhia B, Tran NL, Chan A, et al. The guanine nucleotide exchange factors trio, Ect2, and Vav3 mediate the invasive behavior of glioblastoma. Am J Pathol. 2008 Dec;173(6):1828–1838. S0002-9440(10)61566-0 [pii].
- Gont A, Daneshmand M, Woulfe J, et al. PREX1 integrates G protein-coupled receptor and phosphoinositide 3-kinase signaling to promote glioblastoma invasion. Oncotarget. 2017 Jan 31;8(5):8559–8573. PubMed PMID: 28051998; eng.
- Jarzynka MJ, Hu B, Hui KM, et al. ELMO1 and Dock180, a bipartite Rac1 guanine nucleotide exchange factor, promote human glioma cell invasion. Cancer Res. 2007 Aug 1;67(15):7203–7211. PubMed PMID: 17671188; PubMed Central PMCID: PMCPMC2867339. eng.
- Feng H, Hu B, Liu KW, et al. Activation of Rac1 by Src-dependent phosphorylation of Dock180(Y1811) mediates PDGFRalpha-stimulated glioma tumorigenesis in mice and humans. J Clin Invest. 2011 Dec;121(12):4670–4684. PubMed PMID: 22080864; PubMed Central PMCID: PMCPMC3223070. eng.
- Wong CY, Wuriyanghan H, Xie Y, et al. Epigenetic regulation of phosphatidylinositol 3,4,5-triphosphate-dependent Rac exchanger 1 gene expression in prostate cancer cells. J Biol Chem. 2011 July 22;286(29):25813–25822. M110.211292 [pii].
- Ryan MB, Finn AJ, Pedone KH, et al. ERK/MAPK signaling drives overexpression of the Rac-GEF, PREX1, in BRAF- and NRAS-mutant melanoma. Mol Cancer Res. 2016 Oct;14(10):1009–1018. PubMed PMID: 27418645; PubMed Central PMCID: PMCPMC5065759. eng.
- Rosmaninho P, Mukusch S, Piscopo V, et al. Zeb1 potentiates genome-wide gene transcription with Lef1 to promote glioblastoma cell invasion. Embo J. 2018 Aug 1;37(15). PubMed PMID: 29903919; PubMed Central PMCID: PMCPMC6068449. eng.
- Singh DK, Kollipara RK, Vemireddy V, et al. Oncogenes activate an autonomous transcriptional regulatory circuit that drives glioblastoma. Cell Rep. 2017 Jan 24;18(4):961–976. PubMed PMID: 28122245; PubMed Central PMCID: PMCPMC5321610. eng.
- Welch HC, Coadwell WJ, Ellson CD, et al. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002 Mar 22;108(6):809–821. S0092867402006633 [pii].
- Welch HC. Regulation and function of P-Rex family Rac-GEFs. Small GTPases. 2015 April 3;6(2):49–70. .
- Lu M, Ravichandran KS. Dock180-ELMO cooperation in Rac activation. Methods Enzymol. 2006;406:388–402. PubMed PMID: 16472672; eng.
- Feng H, Hu B, Jarzynka MJ, et al. Phosphorylation of dedicator of cytokinesis 1 (Dock180) at tyrosine residue Y722 by Src family kinases mediates EGFRvIII-driven glioblastoma tumorigenesis. Proc Natl Acad Sci U S A. 2012 Feb 21;109(8):3018–3023. PubMed PMID: 22323579; PubMed Central PMCID: PMCPMC3286964. eng.
- Cote JF, Motoyama AB, Bush JA, et al. A novel and evolutionarily conserved PtdIns(3,4,5)P3-binding domain is necessary for DOCK180 signalling. Nat Cell Biol. 2005 Aug;7(8):797–807. PubMed PMID: 16025104; PubMed Central PMCID: PMCPMC1352170. eng.
- Laurin M, Cote JF. Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes Dev. 2014 Mar 15;28(6):533–547. . PubMed PMID: 24637113; PubMed Central PMCID: PMCPMC3967044. eng.
- Gadea G, Blangy A. Dock-family exchange factors in cell migration and disease. Eur J Cell Biol. 2014 Oct;93(10–12):466–477. . PubMed PMID: 25022758; eng.
- Schmidt S, Debant A. Function and regulation of the Rho guanine nucleotide exchange factor Trio. Small GTPases. 2014;5:e29769. PubMed PMID: 24987837; PubMed Central PMCID: PMCPMC4114922. eng.
- Torrino S, Visvikis O, Doye A, et al. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev Cell. 2011 Nov 15;21(5):959–965. PubMed PMID: 22036506; eng.
- Oberoi TK, Dogan T, Hocking JC, et al. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. Embo J. 2012 Jan 4;31(1):14–28. PubMed PMID: 22117219; PubMed Central PMCID: PMCPMC3252583. eng.
- Zhao J, Mialki RK, Wei J, et al. SCF E3 ligase F-box protein complex SCF(FBXL19) regulates cell migration by mediating Rac1 ubiquitination and degradation. FASEB J. 2013 Jul;27(7):2611–2619. PubMed PMID: 23512198; PubMed Central PMCID: PMCPMC3688740. eng.
- Zhang L, Anglesio MS, O‘Sullivan M, et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat Med. 2007 Sep;13(9):1060–1069. PubMed PMID: 17694067; eng.
- Bouzelfen A, Alcantara M, Kora H, et al. HACE1 is a putative tumor suppressor gene in B-cell lymphomagenesis and is down-regulated by both deletion and epigenetic alterations. Leuk Res. 2016 Jun;45:90–100. PubMed PMID: 27107267; eng.
- Kucuk C, Hu X, Iqbal J, et al. HACE1 is a tumor suppressor gene candidate in natural killer cell neoplasms. Am J Pathol. 2013 Jan;182(1):49–55. PubMed PMID: 23142381; PubMed Central PMCID: PMCPMC3532710. eng.
- Acosta MI, Urbach S, Doye A, et al. Group-I PAKs-mediated phosphorylation of HACE1 at serine 385 regulates its oligomerization state and Rac1 ubiquitination. Sci Rep. 2018 Jan 23;8(1):1410. PubMed PMID: 29362425; PubMed Central PMCID: PMCPMC5780496. eng.
- Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000 Jan 20;403(6767):339–342. . PubMed PMID: 10659857; eng.
- Castillo-Lluva S, Tatham MH, Jones RC, et al. SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nat Cell Biol. 2010 Nov;12(11):1078–1085. PubMed PMID: 20935639; PubMed Central PMCID: PMCPMC2992316. eng.
- Graziano BR, Gong D, Anderson KE, et al. A module for Rac temporal signal integration revealed with optogenetics. J Cell Biol. 2017 Aug 7;216(8):2515–2531. PubMed PMID: 28687663; PubMed Central PMCID: PMCPMC5551696. eng.
- Krause M, Gautreau A. Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat Rev Mol Cell Biol. 2014 Sep;15(9):577–590. PubMed PMID: 25145849; eng.
- Hoshino D, Branch KM, Weaver AM. Signaling inputs to invadopodia and podosomes. J Cell Sci. 2013 Jul 15;126(Pt 14):2979–2989. . PubMed PMID: 23843616; PubMed Central PMCID: PMCPMC3711196. eng.
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998 Jan 23;279(5350):509–514. PubMed PMID: 9438836; eng.
- Kobayashi K, Kuroda S, Fukata M, et al. p140Sra-1 (specifically Rac1-associated protein) is a novel specific target for Rac1 small GTPase. J Biol Chem. 1998 Jan 2;273(1):291–295. PubMed PMID: 9417078; eng.
- Chen Z, Borek D, Padrick SB, et al. Structure and control of the actin regulatory WAVE complex. Nature. 2010 Nov 25;468(7323):533–538. PubMed PMID: 21107423; PubMed Central PMCID: PMCPMC3085272. eng.
- Chen B, Chou HT, Brautigam CA, et al. Rac1 GTPase activates the WAVE regulatory complex through two distinct binding sites. eLife. 2017 Sep 26;6. PubMed PMID: 28949297; PubMed Central PMCID: PMCPMC5614565. eng.
- Law AL, Vehlow A, Kotini M, et al. Lamellipodin and the Scar/WAVE complex cooperate to promote cell migration in vivo. J Cell Biol. 2013 Nov 25;203(4):673–689. PubMed PMID: 24247431; PubMed Central PMCID: PMCPMC3840943. eng.
- Dang I, Gorelik R, Sousa-Blin C, et al. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature. 2013 Nov 14;503(7475):281–284. PubMed PMID: 24132237; eng.
- Wheeler AP, Wells CM, Smith SD, et al. Rac1 and Rac2 regulate macrophage morphology but are not essential for migration. J Cell Sci. 2006 Jul 1;119(Pt 13):2749–2757. PubMed PMID: 16772332; eng.
- Zhuge Y, Xu J. Rac1 mediates type I collagen-dependent MMP-2 activation. role in cell invasion across collagen barrier. J Biol Chem. 2001 May 11;276(19):16248–16256. PubMed PMID: 11340084; eng.
- Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995 Sep 1;269(5228):1270–1272. PubMed PMID: 7652575; eng.
- Perona R, Montaner S, Saniger L, et al. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997 Feb 15;11(4):463–475. PubMed PMID: 9042860; eng.
- Joyce D, Bouzahzah B, Fu M, et al. Integration of Rac-dependent regulation of cyclin D1 transcription through a nuclear factor-kappaB-dependent pathway. J Biol Chem. 1999 Sep 3;274(36):25245–25249. PubMed PMID: 10464245; eng.
- Matos P, Jordan P. Expression of Rac1b stimulates NF-kappaB-mediated cell survival and G1/S progression. Exp Cell Res. 2005 May 1;305(2):292–299. . PubMed PMID: 15817154; eng.
- Tobar N, Caceres M, Santibanez JF, et al. RAC1 activity and intracellular ROS modulate the migratory potential of MCF-7 cells through a NADPH oxidase and NFkappaB-dependent mechanism. Cancer Lett. 2008 Aug 18;267(1):125–132. PubMed PMID: 18433991; eng.
- Nishida K, Kaziro Y, Satoh T. Anti-apoptotic function of Rac in hematopoietic cells. Oncogene. 1999 Jan 14;18(2):407–415. . PubMed PMID: 9927197; eng.
- Pervaiz S, Cao J, Os C, et al. Activation of the RacGTPase inhibits apoptosis in human tumor cells. Oncogene. 2001 Sep 27;20(43):6263–6268. PubMed PMID: 11593437; eng.
- Senger DL, Tudan C, Guiot MC, et al. Suppression of Rac activity induces apoptosis of human glioma cells but not normal human astrocytes. Cancer Res. 2002 Apr 1;62(7):2131–2140. PubMed PMID: 11929835; eng.
- Benitah SA, Frye M, Glogauer M, et al. Stem cell depletion through epidermal deletion of Rac1. Science. 2005 Aug 5;309(5736):933–935. PubMed PMID: 16081735; eng.
- Frances D, Sharma N, Pofahl R, et al. A role for Rac1 activity in malignant progression of sebaceous skin tumors. Oncogene. 2015 Oct;34(43):5505–5512. PubMed PMID: 25659584; eng.
- Yoon CH, Hyun KH, Kim RK, et al. The small GTPase Rac1 is involved in the maintenance of stemness and malignancies in glioma stem-like cells. FEBS Lett. 2011 Jul 21;585(14):2331–2338. PubMed PMID: 21704033; eng.
- Almiron Bonnin DA, Havrda MC, Lee MC, et al. Secretion-mediated STAT3 activation promotes self-renewal of glioma stem-like cells during hypoxia. Oncogene. 2018 Feb 22;37(8):1107–1118. PubMed PMID: 29155422; PubMed Central PMCID: PMCPMC5851110. eng.
- Kumar R, Sanawar R, Li X, et al. Structure, biochemistry, and biology of PAK kinases. Gene. 2017 Mar 20;605:20–31. PubMed PMID: 28007610; PubMed Central PMCID: PMCPMC5250584. eng.
- Advani SJ, Camargo MF, Seguin L, et al. Kinase-independent role for CRAF-driving tumour radioresistance via CHK2. Nat Commun. 2015 Sep 03;6:8154. PubMed PMID: 26333361; PubMed Central PMCID: PMCPMC4559870. eng.
- Li DQ, Nair SS, Ohshiro K, et al. MORC2 signaling integrates phosphorylation-dependent, ATPase-coupled chromatin remodeling during the DNA damage response. Cell Rep. 2012 Dec 27;2(6):1657–1669. PubMed PMID: 23260667; PubMed Central PMCID: PMCPMC3554793. eng.
- Kohn AD, Summers SA, Birnbaum MJ, et al. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996 Dec 6;271(49):31372–31378. PubMed PMID: 8940145; eng.
- Hu H, Juvekar A, Lyssiotis CA, et al. Phosphoinositide 3-kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell. 2016 Jan 28;164(3):433–446. PubMed PMID: 26824656; PubMed Central PMCID: PMCPMC4898774. eng.
- Marin-Valencia I, Yang C, Mashimo T, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012 Jun 6;15(6):827–837. PubMed PMID: 22682223; PubMed Central PMCID: PMCPMC3372870. eng.
- Maher EA, Marin-Valencia I, Bachoo RM, et al. Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed. 2012 Nov;25(11):1234–1244. PubMed PMID: 22419606; PubMed Central PMCID: PMCPMC3406255. eng.
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009 May 22;324(5930):1029–1033. . PubMed PMID: 19460998; PubMed Central PMCID: PMCPMC2849637. eng.
- Miyano K, Sumimoto H. Role of the small GTPase Rac in p22phox-dependent NADPH oxidases. Biochimie. 2007 Sep;89(9):1133–1144. . PubMed PMID: 17583407; eng.
- Cetinbas N, Daugaard M, Ar M, et al. Loss of the tumor suppressor Hace1 leads to ROS-dependent glutamine addiction. Oncogene. 2015 Jul 23;34(30):4005–4010. PubMed PMID: 25284589; PubMed Central PMCID: PMCPMC4387113. eng.
- Hardee ME, Zagzag D. Mechanisms of glioma-associated neovascularization. Am J Pathol. 2012 Oct;181(4):1126–1141. . PubMed PMID: 22858156; PubMed Central PMCID: PMCPMC3463636. eng.
- Bid HK, Roberts RD, Manchanda PK, et al. RAC1: an emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol Cancer Ther. 2013 Oct;12(10):1925–1934. PubMed PMID: 24072884; PubMed Central PMCID: PMCPMC3823055. eng.
- Vader P, van der Meel R, Symons MH, et al. Examining the role of Rac1 in tumor angiogenesis and growth: a clinically relevant RNAi-mediated approach. Angiogenesis. 2011 Dec;14(4):457–466. PubMed PMID: 21789714; eng.
- D‘Amico G, Robinson SD, Germain M, et al. Endothelial-Rac1 is not required for tumor angiogenesis unless alphavbeta3-integrin is absent. PLoS One. 2010 Mar 22;5(3):e9766. PubMed PMID: 20339539; PubMed Central PMCID: PMCPMC2842301. eng.
- Buerki RA, Chheda ZS, Okada H. Immunotherapy of primary brain tumors: facts and hopes. Clin Cancer Res. 2018 Jun 5. PubMed PMID: 29871908; eng. DOI:10.1158/1078-0432.ccr-17-2769.
- Engler JR, Robinson AE, Smirnov I, et al. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS One. 2012;7(8):e43339. PubMed PMID: 22937035; PubMed Central PMCID: PMCPMC3425586. eng.
- Woroniecka KI, Rhodin KE, Chongsathidkiet P, et al. T-cell dysfunction in glioblastoma: applying a new framework. Clin Cancer Res. 2018 Aug 15;24(16):3792–3802. PubMed PMID: 29593027; PubMed Central PMCID: PMCPMC6095741. eng.
- Chongsathidkiet P, Jackson C, Koyama S, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. 2018 Sep;24(9):1459–1468. PubMed PMID: 30104766; PubMed Central PMCID: PMCPMC6129206. eng.
- Ferguson SD, Srinivasan VM, Heimberger AB. The role of STAT3 in tumor-mediated immune suppression. J Neurooncol. 2015 Jul;123(3):385–394. . PubMed PMID: 25700834; eng.
- Nefedova Y, Huang M, Kusmartsev S, et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol. 2004 Jan 1;172(1):464–474. PubMed PMID: 14688356; eng.
- Corzo CA, Cotter MJ, Cheng P, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009 May 1;182(9):5693–5701. PubMed PMID: 19380816; PubMed Central PMCID: PMCPMC2833019. eng.
- Wu A, Wei J, Kong LY, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010 Nov;12(11):1113–1125. PubMed PMID: 20667896; PubMed Central PMCID: PMCPMC3098021. eng.
- Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005 Dec;11(12):1314–1321. PubMed PMID: 16288283; eng.
- Raptis L, Arulanandam R, Geletu M, et al. The R(h)oads to Stat3: stat3 activation by the Rho GTPases. Exp Cell Res. 2011 Aug 1;317(13):1787–1795. PubMed PMID: 21619876; PubMed Central PMCID: PMCPMC3129747. eng.
- Simon AR, Vikis HG, Stewart S, et al. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science. 2000 Oct 6;290(5489):144–147. PubMed PMID: 11021801; eng.
- Faruqi TR, Gomez D, Bustelo XR, et al. Rac1 mediates STAT3 activation by autocrine IL-6. Proc Natl Acad Sci U S A. 2001 Jul 31;98(16):9014–9019. PubMed PMID: 11470914; PubMed Central PMCID: PMCPMC55365. eng.
- Pelletier S, Duhamel F, Coulombe P, et al. Rho family GTPases are required for activation of Jak/STAT signaling by G protein-coupled receptors. Mol Cell Biol. 2003 Feb;23(4):1316–1333. PubMed PMID: 12556491; PubMed Central PMCID: PMCPMC141129. eng.
- Maldonado MDM, Dharmawardhane S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018 Jun 15;78(12):3101–3111. . PubMed PMID: 29858187; PubMed Central PMCID: PMCPMC6004249. eng.
- Marei H, Malliri A. Rac1 in human diseases: the therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases. 2017 Jul 3;8(3):139–163. . PubMed PMID: 27442895; PubMed Central PMCID: PMCPMC5584733. eng.
- Desire L, Bourdin J, Loiseau N, et al. RAC1 inhibition targets amyloid precursor protein processing by gamma-secretase and decreases Abeta production in vitro and in vivo. J Biol Chem. 2005 Nov 11;280(45):37516–37525. PubMed PMID: 16150730; eng.
- Shutes A, Onesto C, Picard V, et al. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007 Dec 7;282(49):35666–35678. PubMed PMID: 17932039; eng.
- Khosravi-Far R, Solski PA, Clark GJ, et al. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol Cell Biol. 1995 Nov;15(11):6443–6453. PubMed PMID: 7565796; PubMed Central PMCID: PMCPMC230895. eng.
- Qiu RG, Chen J, Kirn D, et al. An essential role for Rac in Ras transformation. Nature. 1995 Mar 30;374(6521):457–459. PubMed PMID: 7700355; eng.
- Weiss WA, Burns MJ, Hackett C, et al. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Res. 2003 Apr 1;63(7):1589–1595. PubMed PMID: 12670909; eng.
- Gao Y, Dickerson JB, Guo F, et al. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004 May 18;101(20):7618–7623. PubMed PMID: 15128949; PubMed Central PMCID: PMCPMC419655. eng.
- Karpel-Massler G, Westhoff MA, Zhou S, et al. Combined inhibition of HER1/EGFR and RAC1 results in a synergistic antiproliferative effect on established and primary cultured human glioblastoma cells. Mol Cancer Ther. 2013 Sep;12(9):1783–1795. PubMed PMID: 23832120; eng.
- Hernandez E, De La Mota-Peynado A, Dharmawardhane S, et al. Novel inhibitors of Rac1 in metastatic breast cancer. P R Health Sci J. 2010 Dec;29(4):348–356. PubMed PMID: 21261173; eng.
- Castillo-Pichardo L, Humphries-Bickley T, De La Parra C, et al. The Rac inhibitor EHop-016 inhibits mammary tumor growth and metastasis in a nude mouse model. Transl Oncol. 2014 Oct;7(5):546–555. PubMed PMID: 25389450; PubMed Central PMCID: PMCPMC4225654. eng.
- Humphries-Bickley T, Castillo-Pichardo L, Hernandez-O‘Farrill E, et al. Characterization of a Dual Rac/Cdc42 Inhibitor MBQ-167 in metastatic cancer. Mol Cancer Ther. 2017 May;16(5):805–818. PubMed PMID: 28450422; PubMed Central PMCID: PMCPMC5418092. eng.
- Cardama GA, Comin MJ, Hornos L, et al. Preclinical development of novel Rac1-GEF signaling inhibitors using a rational design approach in highly aggressive breast cancer cell lines. Anticancer Agents Med Chem. 2014;14(6):840–851. PubMed PMID: 24066799; PubMed Central PMCID: PMCPMC4104455. eng.
- Cardama GA, Gonzalez N, Ciarlantini M, et al. Proapoptotic and antiinvasive activity of Rac1 small molecule inhibitors on malignant glioma cells. Onco Targets Ther. 2014;7:2021–2033. PubMed PMID: 25378937; PubMed Central PMCID: PMCPMC4218912. eng.
- Vigil D, Cherfils J, Rossman KL, et al. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010 Dec;10(12):842–857. PubMed PMID: 21102635; PubMed Central PMCID: PMCPMC3124093. eng.
- Vives V, Laurin M, Cres G, et al. The Rac1 exchange factor Dock5 is essential for bone resorption by osteoclasts. J Bone Miner Res. 2011 May;26(5):1099–1110. PubMed PMID: 21542010; PubMed Central PMCID: PMCPMC4640905. eng.
- Ferrandez Y, Zhang W, Peurois F, et al. Allosteric inhibition of the guanine nucleotide exchange factor DOCK5 by a small molecule. Sci Rep. 2017 Oct 31;7(1):14409. PubMed PMID: 29089502; PubMed Central PMCID: PMCPMC5663973. eng.
- Hill K, Krugmann S, Andrews SR, et al. Regulation of P-Rex1 by phosphatidylinositol (3,4,5)-trisphosphate and Gbetagamma subunits. J Biol Chem. 2005 Feb 11;280(6):4166–4173. M411262200 [pii].
- Zhao HF, Wang J, Shao W, et al. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: current preclinical and clinical development. Mol Cancer. 2017 Jun 7;16(1):100. PubMed PMID: 28592260; PubMed Central PMCID: PMCPMC5463420. eng.
- Rane CK, Minden A. P21 activated kinase signaling in cancer. Semin Cancer Biol. 2018 Jan 9. PubMed PMID: 29330094; eng. DOI:10.1016/j.semcancer.2018.01.006.
- Licciulli S, Maksimoska J, Zhou C, et al. FRAX597, a small molecule inhibitor of the p21-activated kinases, inhibits tumorigenesis of neurofibromatosis type 2 (NF2)-associated Schwannomas. J Biol Chem. 2013 Oct 4;288(40):29105–29114. PubMed PMID: 23960073; PubMed Central PMCID: PMCPMC3790009. eng.
- Chow HY, Jubb AM, Koch JN, et al. p21-Activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 2012 Nov 15;72(22):5966–5975. PubMed PMID: 22983922; PubMed Central PMCID: PMCPMC3500416. eng.
- Dolan BM, Duron SG, Campbell DA, et al. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc Natl Acad Sci U S A. 2013 Apr 2;110(14):5671–5676. PubMed PMID: 23509247; PubMed Central PMCID: PMCPMC3619302. eng.
- Siekmann IK, Dierck K, Prall S, et al. Combined inhibition of receptor tyrosine and p21-activated kinases as a therapeutic strategy in childhood ALL. Blood Adv. 2018 Oct 9;2(19):2554–2567. PubMed PMID: 30301811; PubMed Central PMCID: PMCPMC6177654. eng.
- Zhou J, Atsina KB, Himes BT, et al. Novel delivery strategies for glioblastoma. Cancer J. 2012 Jan -Feb;18(1):89–99. PubMed PMID: 22290262; PubMed Central PMCID: PMCPMC3269656. eng.
- Zeghouf M, Guibert B, Zeeh JC, et al. Arf, Sec7 and Brefeldin A: a model towards the therapeutic inhibition of guanine nucleotide-exchange factors. Biochem Soc Trans. 2005 Dec;33(Pt 6):1265–1268. PubMed PMID: 16246094; eng.
- Zeeh JC, Zeghouf M, Grauffel C, et al. Dual specificity of the interfacial inhibitor brefeldin a for arf proteins and sec7 domains. J Biol Chem. 2006 Apr 28;281(17):11805–11814. PubMed PMID: 16484231; eng.