
Abstract
Osteoarthritis (OA) poses a growing threat to the health of the global population. Accumulation of advanced glycation end-products (AGEs) has been shown to upregulate expression of degradative enzymes such as matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) in chondrocytes, which leads to excessive degradation of type II collagen and aggrecan in the articular extracellular matrix (ECM). In the present study we investigated the effects of the GLP-1 agonist lixisenatide, a widely used type II diabetes medication, on AGEs-induced decreased mitochondrial membrane potential (MMP), degradation of ECM, oxidative stress, expression of cytokines including interleukin (IL)-1β and IL-6, and activation of nuclear factor kappa B (NF-κB). Our findings indicate that lixisenatide significantly ameliorated the deleterious effects of AGEs in a dose-dependent manner. Thus, lixisenatide has potential as a safe and effective treatment for OA and other AGEs-induced inflammatory diseases.
Introduction
Osteoarthritis (OA) is one of the world’s leading debilitating age-related diseases and is expected to become more than twice as prevalent over the next decade. In large part, this is due to an increase in the average age of the global population owing to advancements in healthcare and lifestyle improvements [Citation1]. Advanced glycation end-products (AGEs) are the byproduct of non-enzymatic protein glycation and accumulate in the body due to their resilience to degradation. Additionally, AGEs are used as a food preservative, thereby also entering the body through diet [Citation2]. Chondrocytes have been shown to express the receptor for AGEs (RAGE), indicating that AGEs play a role in regular cell turnover and cartilage remodeling [Citation3]. It has also been shown that accumulation of AGEs promotes the release of pro-inflammatory cytokines including tumor necrosis factor alpha (TNF-α) and interleukins (ILs), activation of nuclear factor kappa B (NF-κB), and production of reactive oxygen species (ROS) [Citation4,Citation5].
The main hallmark of OA is the excessive degradation of the articular extracellular matrix (ECM). In joint cartilage, the ECM is primarily composed of type II collagen and aggrecan and accounts for roughly 95% of the cartilage tissue mass. Of these, type II collagen is fibrillar collagen with rigid and stress-resilient properties that provide cartilage with its high degree of structural integrity [Citation6]. Owing to its resilience to degradation under normal physiological conditions, type II collagen has a very slow rate of cell turnover. Therefore, excessive degradation of type II collagen is largely considered to be irreversible. Matrix metalloproteinases (MMPs) are a family of zinc-dependent enzymes that target type II collagen for degradation. In normal physiological conditions, MMPs play important roles in morphogenesis, embryonic development, tissue repair, and the inflammatory disease process [Citation7–9]. Of these, MMP-3 (stromelysin-1) and MMP-13 (collagenase 3) have been cited as the main collagenases responsible for cleavage of type II collagen by unwinding the collagen triple helix and cleaving the P4-P11 site [Citation10]. Aggrecan is the most abundant proteoglycan in articular cartilage and allows joints to withstand the compressive force and absorb shock [Citation11]. Loss of aggrecan is an early event in the pathogenesis of OA and expression of cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin (IL)-6 has been shown to induce cleavage of aggrecans by upregulating the expression of a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) [Citation12]. The aggrecanases ADAMTS-4 and ADAMTS-5 cleave aggrecan to promote regular cell turnover and homeostasis, however, increased expression of ADAMTS-4 and ADAMTS-5 has been shown to be involved in the destruction of aggrecan in a human OA model [Citation13]. However, in the pathological condition of OA, increased secretion of IL-6 and TNF-α leads to enhanced expression of MMPs and ADAMTS, thereby resulting in excessive and irreversible degradation of the ECM [Citation14]. Additionally, overproduction of reactive oxygen species (ROS) and activation of nuclear factor kappa B (NF-κB) have been shown to play a role in OA. ROS are the byproduct of numerous physiological processes and play important roles in normal homeostasis; however, overproduction of ROS promotes expression of MMPs, ADAMTS, and exacerbates generation of ROS, resulting in increased and sustained degradation of the ECM. Furthermore, it has been shown that AGEs directly influence the generation of ROS through catalytic sites in their molecular structure [Citation15]. Activation of the NF-κB pro-inflammatory pathway through phosphorylation of nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκBα) further induces the inflammatory response and upregulates expression of MMPs and ADAMTS, thereby promoting the progression of OA [Citation16].
Lixisenatide is an agonist of the receptor for glucagon-like peptide 1 (GLP-1R), an endogenous incretin hormone responsible for mediating the secretion of glucagon [Citation17]. Lixisenatide is widely used to increase the release of insulin by β-cells in the treatment of type II diabetes. Lixisenatide has displayed a wide range of pharmacological properties in different kinds of diseases. Mouse model experiments have demonstrated the anti-inflammatory effects of lixisenatide. For example, in a murine model of Alzheimer’s disease, lixisenatide was shown to exert neuroprotective effects by reducing synapse loss and preventing chronic inflammation [Citation18]. In a recent study involving atherogenic-diet-fed Apoe −/− Irs2 +/− mice, lixisenatide was shown to exert anti-inflammatory and antiatherogenic effects by downregulating expression of IL-6 and discouraging macrophages from shifting to the M2 proinflammatory phenotype [Citation19]. Interestingly, a recent literature review showed that GLP-1 agonists such as lixisenatide exert anti-inflammatory effects in a wide range of disease including types 1 and 2 diabetes, atherosclerosis, neurodegenerative disorders, nonalcoholic steatohepatitis, diabetic nephropathy, asthma, and psoriasis [Citation20]. However, there is still little research regarding the role of GLP-1 in bone homeostasis. A recent review investigating the role of GLP-1 in bone metabolism determined that GLP-1 may regulate bone formation and bone structure, but the existing research is not adequate to confirm this [Citation21]. In the present study, we investigated the effects of agonism of GLP-1R using lixisenatide on AGEs-induced degradation of the ECM. Our findings demonstrate that lixisenatide has potential as a novel treatment against OA by rescuing AGEs-induced mitochondrial dysfunction, ameliorating oxidative stress, preventing degradation of type II collagen and aggrecan through downregulation of MMP-3, MMP-13, ADAMTS-4 and ADAMTS-5, exerting an anti-inflammatory effect by reducing expression of TNF-α and IL-6, and inhibiting activation of the proinflammatory NF-κB pathway by preventing phosphorylation of IκBα.
Materials and methods
Cartilage explant cultures
Human primary chondrocytes (HPCs) were commercially purchased from MT-BIO Company (China). The cells were cultured in DMEM/Ham’s F-12 medium (Thermo Fisher Scientific, USA) containing 10% FBS, 100 μg/ml streptomycin, and 100 IU/ml penicillin. A portion of the cells was frozen in a liquid nitrogen tank for later use, and the remaining cells were treated with 100 μg/ml AGEs in the presence or absence of 10 and 20 nM lixisenatide for 48 h.
Western blot
Treated and non-treated chondrocytes were washed with PBS for protein extraction using cell lysis buffer (Cell Signaling, USA) containing protease and phosphatase inhibitors. Equal amounts (20–50 μg) of total protein or nuclear protein were loaded onto 4–12% Mini-PROTEAN® TBE Precast Gels (Bio-Rad, USA). Protein in the acrylamide gel was transferred onto a PVDF membrane (Thermo Fisher Scientific, USA). The membrane was then blocked in 5% slim milk diluted in TBS containing 0.1% Tween 20 (TBS-T) for 1 h. The membrane was incubated overnight with primary antibody at 4 °C, rinsed three times in TBS-T, and incubated with secondary antibody for 1 h at room temperature (RT). Detection of immunoreactive bands was performed using chemiluminescence (Novex ECL, Invitrogen).
Real-time polymerase chain reaction (RT-PCR)
Total RNA was isolated from human chondrocytes in accordance with the manufacturer’s instructions for the use of TRIzol reagent (Thermo Fisher Scientific, USA). Total RNA was reverse-transcribed to cDNA using an iScript cDNA synthesis kit (Bio-Rad). Quantitative PCR was performed using Advanced SYBR Green Supermix (Bio-Rad) and amplified on a StepOne Plus real-time PCR system (Applied Science) to obtain cycle threshold (Ct) values for target and internal reference cDNA levels.
Preparation of nuclear extracts from chondrocytes
Nuclear protein was extracted from human primary chondrocytes using a commercial kit (Thermo Fisher Scientific, USA). Nuclear protein concentrations were determined by the BCA method. The nuclear protein lamin B1 was used as an internal control. Nuclear levels of NF-κB p65 were examined using western blot analysis to determine NF-κB activation.
Measurement of 4-hydroxy-2-nonenal (4-HNE) immunofluorescence
Intracellular levels of 4-HNE in chondrocytes were measured using the immunofluorescence method. Firstly, chondrocytes were fixed with 4% paraformaldehyde for 15 min at RT and then permeabilized with 0.4% Triton X-100 for 10 min. After blocking with 5% BSA and 2.5% FBS, cells were incubated with the primary anti-4HNE antibody (ab48506, Abcam, USA) for 2 h at RT. After three washes, cells were probed with Alexa-488 conjugated secondary antibodies (Invitrogen, USA) for another 1 h at RT. Fluorescent signals were visualized with an inverted fluorescence microscope (Zeiss, Germany).
Determination of mitochondrial membrane potential (MMP)
Intracellular MMP in chondrocytes was assessed with tetramethylrhodamine methyl ester (TMRM). Cells were treated with 100 μg/ml AGEs in the presence or absence of 10 and 20 nM lixisenatide for 48 h. After 3 washes with PBS, cells were incubated with 20 nmol/L TMRM for 30 min at 37 °C in darkness. Fluorescent signals were visualized with an inverted fluorescence microscope (Zeiss, Germany).
Measurement of adenosine triphosphate (ATP) levels via bioluminescence assay
The level of intracellular ATP in chondrocytes was measured using an ATP bioluminescence assay kit (#A22066, Thermo Fisher Scientific, USA). Briefly, chondrocytes were lysed with the lysis buffer included in the kit and centrifuged at 10,000 × g. Supernatant (100 μl) was mixed with another 10 μl luciferin/luciferase reagent. Light output was recorded with a microplate luminometer.
ELISA assay
Secretion of TNF-α and IL-6 from human primary chondrocytes into the cell culture medium was measured using a commercial ELISA kit obtained from R&D Systems in accordance with the manufacturer’s protocols.
Luciferase reporter gene assay
To determine NF-κB transcriptional activity, NF-κB promoter-luciferase (Clontech, USA) and β-galactosidase plasmid were transfected into chondrocytes using Lipofectamine 2000. Human primary chondrocytes were treated with 100 μg/ml AGEs in the presence or absence of 10 and 20 nM lixisenatide for 48 h. Cells were then lysed to measure luciferase activity and β-galactosidase activity using a commercial dual luminescence assay kit (Gene Copoeia, MD) with a luminometer. Luciferase activity was normalized to β-galactosidase activity.
Statistical analysis
All data are presented as means ± SEM Statistical analyses were performed using analysis of variance (ANOVA). p-values < .05 were considered statistically significant.
Results
Rescue of AGEs-induced mitochondrial dysfunction
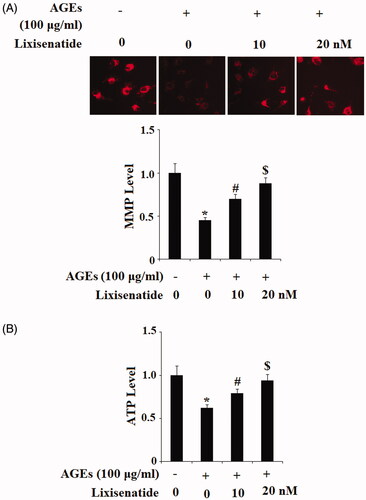
Mitochondrial dysfunction induced by the presence of AGEs has been observed in age-related diseases including OA. To determine the effects of lixisenatide on AGEs-induced mitochondrial dysfunction, we assessed mitochondrial membrane potential (MMP) and production of adenosine triphosphate (ATP) using TMRM and a luciferase assay. Briefly, human primary chondrocytes were treated with 100 μg/ml AGEs in the presence or absence of 10 and 20 nM lixisenatide for 48 h. As shown in , exposure to AGEs drastically lowered the level of MMP by more than half, which was restored by treatment with lixisenatide in a dose-dependent manner. Notably, 20 µg lixisenatide almost completely restored MMP to basal levels. As shown in , these effects were confirmed by the results of luciferase assay showing that exposure to AGEs reduced mitochondrial ATP production by nearly half, which was rescued by treatment with lixisenatide. Again, 20 µg lixisenatide almost completely ameliorated the effects of AGEs on ATP level.
Figure 1. Lixisenatide ameliorates advanced glycation end products (AGEs)-induced mitochondrial dysfunction. (A) Mitochondrial membrane potential was measured by TMRM; (B) Intracellular ATP levels were determined by a luciferase assay (*, #, $, p < .01).
Reduction of AGEs-induced oxidative stress
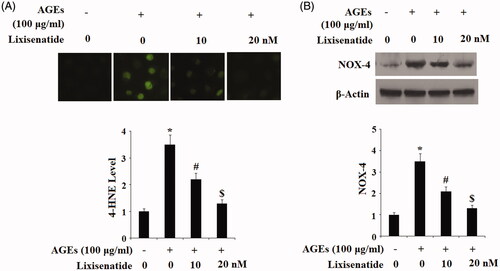
Oxidative stress plays a major role in chronic inflammation. The α, β-unsaturated hydroxyalkenal 4-hydroxy-2-nonenal (HNE-4) is a byproduct of lipid peroxidation. NADPH oxidase 4 (NOX4) has been shown to protect against inflammatory stress and is upregulated in response to oxidative stress. To investigate the effects of lixisenatide on AGEs-induced oxidative stress, HPCs were again exposed to 100 µg AGEs in the presence or absence of 10 and 30 µM lixisenatide for 48 h. As shown in , exposure to AGEs elevated expression of 4-HNE by approximately 3.5-fold, which was ameliorated by lixisenatide in a dose-dependent manner. The results of western blot analysis in demonstrate that exposure to AGEs drastically increased protein expression of NOX4, which was also ameliorated by lixisenatide in a dose-dependent manner. Notably, a dose of 20 µg lixisenatide again rescued expression of 4-HNE and NOX4 to near basal levels, indicating that lixisenatide may possess a protective effect against oxidative stress in HPCs.
Figure 2. Lixisenatide ameliorates advanced glycation end products (AGEs)-induced oxidative stress. (A) The lipid peroxidation 4-HNE was determined by immunostaining; (B). NOX-4 was determined by western blot analysis (*, #, $, p < .01).
Reduction of AGEs-induced expression of MMPs and degradation of type II collagen
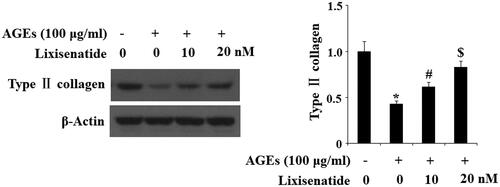
Degradation of type II collagen by MMP-3 and MMP-13 is a major event in the pathogenesis of OA. Here, we tested the effects of treatment with 10 and 20 µM lixisenatide on HPCs exposed to 100 µg AGEs for 48 h. As demonstrated by the results in , exposure to AGEs caused a significant elevation in the expression of MMP-3 and MMP-13, which was significantly prevented by lixisenatide at both the mRNA and protein levels in a dose-dependent manner. Concordantly, the results in show that treatment with 10 and 20 µM lixisenatide also rescued AGEs-mediated degradation of type II collagen in HPCs at both the mRNA and protein levels in a dose-dependent manner.
Figure 3. Lixisenatide ameliorates advanced glycation end products (AGEs)-induced expression of MMP-3 and MMP-13. (A) Expression of MMP-3 and MMP-13 determined by real-time PCR analysis; (B) Expression of MMP-3 and MMP-13 determined by western blot analysis (*, #, $, p < .01).
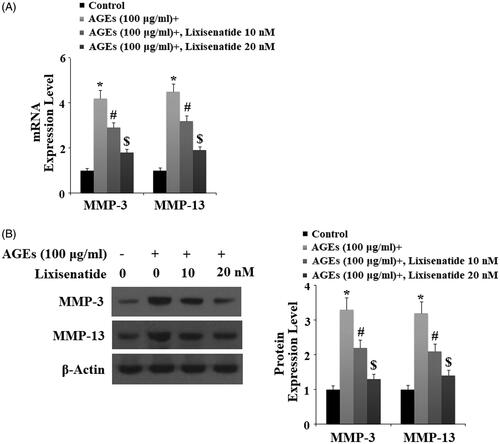
Figure 4. Lixisenatide ameliorates advanced glycation end products (AGEs)-induced degradation of type II collagen. Type II collagen was determined by western blot analysis (*, #, $, p < .01).
Reduction of AGEs-induced expression of ADAMTS and degradation of aggrecan
ADAMTS-4 and ADAMTS-5 are the major aggrecanases responsible for degradation of aggrecan in the articular ECM. To investigate the effects of clinically relevant doses of lixisenatide on AGEs-induced expression of ADAMTS-4 and ADAMTS-5, we exposed HPCs to 100 µg AGEs in the presence or absence of 10 and 20 µM lixisenatide for 48 h. As shown by the results of real-time PCR and western blot analysis in , exposure to AGEs drastically increased expression of ADAMTS-4 and ADAMTS-5 and both the mRNA and protein levels, respectively. Again, 20 µM lixisenatide almost completely ameliorated AGEs-induced elevated levels of ADAMTS-4 and ADAMTS-5. To further confirm the protective effect of lixisenatide against AGEs-induced degradation of aggrecan through downregulation of ADAMTS expression, we investigated the degradation of aggrecan through western blot analysis. The results in confirm that treatment with lixisenatide prevented degradation of aggrecan induced by AGEs.
Figure 5. Lixisenatide ameliorates advanced glycation end products (AGEs)-induced expression of ADAMTS-4 and ADAMTS-5. (A) Expression of ADAMTS-4 and ADAMTS-5 were determined by real-time PCR analysis; (B) Expression of ADAMTS-4 and ADAMTS-5 were determined by western blot analysis (*, #, $, p < .01).
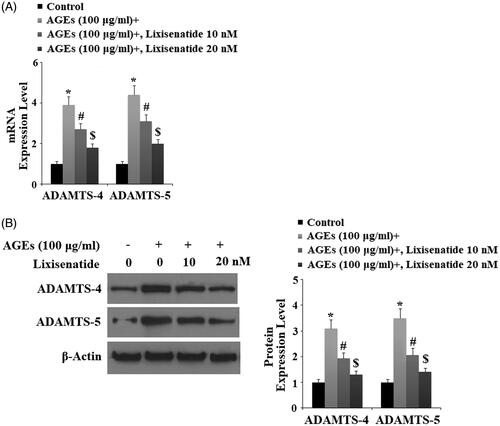
Figure 6. Lixisenatide ameliorates advanced glycation end products (AGEs)-induced degradation of aggrecan. Aggrecan was determined by western blot analysis (*, #, $, p < .01).
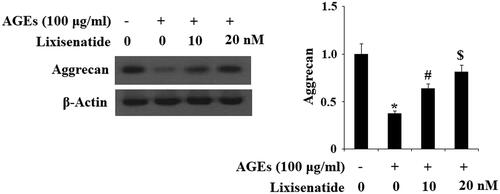
Reduced expression of proinflammatory cytokines
Proinflammatory cytokines such as TNF-α and IL-6 have been shown to play a pivotal role in the development and progression of OA. Accumulation of AGEs elevates expression of TNF-α and IL-6, thereby sustaining the inflammatory response. To investigate the effects of lixisenatide on AGEs-induced expression of these cytokines, HPCs were exposed to 100 µg AGEs for 48 h in the presence or absence of 10 and 20 µM lixisenatide to induce expression of TNF-α and IL-6. As shown by the results of real-time PCR and western blot analysis in , exposure to AGEs increased expression of TNF-α and IL-6 by roughly 5- and 4-fold, respectively, at both the mRNA and protein levels. However, the expression of both of these cytokines was prevented by treatment with lixisenatide in a dose-dependent manner.
Figure 7. Lixisenatide ameliorates advanced glycation end products (AGEs)-induced secretion of TNF-α and IL-6. (A) Expression of TNF-α and IL-6 at the gene level was determined real-time PCR analysis; (B) Secretion of TNF-α and IL-6 at the protein level was determined ELISA (*, #, $, p < .01).
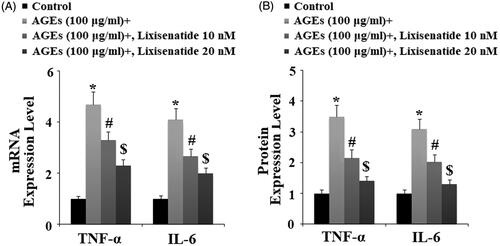
Reduction of NF-κB via rescue of phosphorylated IκBα
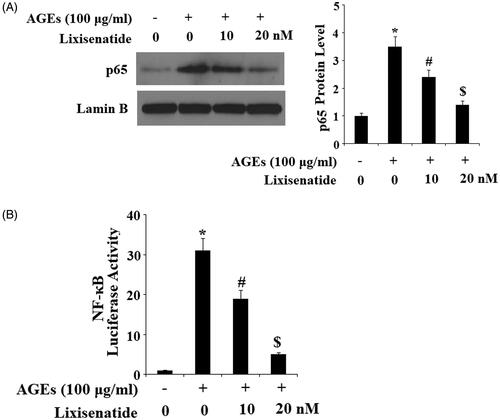
NF-κB is a major regulator of inflammation. Under normal conditions, NF-κB is sequestered in the cytoplasm by its inhibitor IκBα. However, under pathological conditions, phosphorylation of IκBα leads to translocation of p65 to nuclei and subsequent activation of NF-κB, thereby driving the inflammatory response. To determine the effects of lixisenatide on AGEs-induced phosphorylation and degradation of IκBα as well as activation of NF-κB, we exposed HPCs to 100 µg AGEs in the presence or absence of 10 and 20 µM lixisenatide for 6 h. As demonstrated by the results of western blot analysis in , exposure to AGEs significantly elevated the level of phosphorylated IκBα and decreased the level of total IκBα. Treatment with lixisenatide successfully reduced these effects of AGEs in a dose-dependent manner. Using lamin-B as a positive control, we next set out to determine the effects of clinically relevant doses of lixisenatide on activation of NF-κB by exposing HPCs to 100 µg AGEs in the presence or absence of lixisenatide for 48 h. As demonstrated by the results in , AGEs significantly increased the nuclear translocation of p65 protein and luciferase activity of NF-κB, which were both ameliorated by treatment with lixisenatide in a dose-dependent manner.
Figure 8. Lixisenatide ameliorates advanced glycation end products (AGEs)-induced phosphorylation and degradation of IκBα. Phosphorylated and total levels of IκBα were determined by western blot analysis (*, #, $, p < .01).
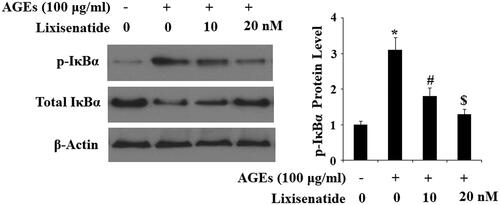
Figure 9. Lixisenatide ameliorates advanced glycation end products (AGEs)-induced activation of NF-κB. (A) Nuclear translocation of p65; lamin B was used as a positive control; (B) Luciferase activity of NF-κB (*, #, $, p < .01).
Discussion
The results of the present study demonstrate that administration of lixisenatide at clinically relevant doses has a beneficial protective effect against multiple aspects of OA, including degradation of the articular ECM, mitochondrial dysfunction, oxidative stress, expression of proinflammatory cytokines, and activation of the NF-κB proinflammatory signaling pathway. Although the exact mechanisms through which lixisenatide exerts these effects remains unclear, as a specific GLP-1R agonist, we can hypothesize that activation of GLP-1R on primary chondrocytes plays a role in mediating the deleterious effects of AGEs as seen in OA. The incretin peptide GLP-1 has been widely studied as a therapeutic target for the treatment of type II diabetes due to its role in promoting insulin secretion by pancreatic β-cells and has been praised for its ability to slow gastric emptying, inhibit glucagon secretion and promote weight loss [Citation22]. Mechanistically, a recent study showed that GLP-1 could inhibit the expression of MMPs, including MMP-3 and MMP-13 [Citation23]. As a safe and effective therapeutic option, the specific GLP-1R agonist lixisenatide was approved by the Food and Drug Administration (FDA) in 2013 and has since been widely used for the treatment of type II diabetes [Citation24]. Thus, we set out to investigate the potential of this drug in preventing AGEs-induced degradation of the articular ECM.
Our results show that consistent with previous studies, agonism of GLP-1R by lixisenatide reduced degradation of type II collagen and aggrecan by collagenases (MMP-3, MMP-13) and aggrecanases (ADAMTS-4, ADAMTS-5). As these are recognized as the most important proteinases in OA, the capacity of GLP-1R to downregulate increased expression of these factors induced by AGEs is of great value as a potential therapeutic target for OA. Additionally, we determined that treatment with lixisenatide can rescue reduced mitochondrial membrane potential (MMP) and ATP production induced by AGEs. Loss of mitochondrial function leads to reduced production of ATP, the body’s main source of energy. Such loss of mitochondrial function has been shown to play a major role in age-related disease due to decreased oxidative phosphorylation through the electron transport chain [Citation25]. The capacity of lixisenatide to ameliorate mitochondrial dysfunction induced by AGEs may be of great value in a wide range of diseases. Oxidative stress also plays a major role in a wide range of disease states. In part, this is because the generation of ROS, NOX4 and sustained oxidative stress has a self-promoting role in chronic inflammatory diseases. That is to say, high levels of oxidative stress further exacerbate generation of ROS and release of 4-HNE and NOX4, which in turn raises the level of oxidative stress.
TNF-α and IL-6 are two of the major proinflammatory cytokines involved in OA. TNF-α has been shown to exacerbate the pathological progression of OA by upregulating generation of MMPs, ADAMTS, ROS and other factors [Citation26,Citation27]. As a class of proinflammatory cytokines, interleukins (ILs) have been shown to play critical roles in a wide range of chronic inflammatory diseases including OA. Of these, the expression of IL-6, which is upregulated by oxidative stress and ROS, has been shown to be involved in the pathogenesis of OA [Citation14,Citation28]. Furthermore, this response is mediated through activation of the NF-κB signaling pathway [Citation29,Citation30]. The NF-κB transcription factor is ubiquitously expressed and plays a key role in triggering and sustaining the inflammatory response. Here, the IKKα/IκBα/NF-κB inflammatory pathway has been shown to mediate inflammation, cytokine production, and cartilage degradation, among other things [Citation16]. Under normal physiological conditions, NF-κB exists in the cytoplasm in its inactive state as a heterodimer of the p50/p65 subunits, which is maintained by IκBα, a member of the IκB class of inhibitory proteins. However, oxidative stress leads to activation of the p38 mitogen-activated protein kinase (MAPK) pathway and subsequent phosphorylation of IκBα by IKKα/β, thereby resulting in nuclear translocation of p65 and activation of NF-κB [Citation31–33]. Our findings show that treatment with lixisenatide exerted a dose-dependent reduction of AGEs-induced expression of IL-6 and TNF-α, activation of the NF-κB signaling pathway, generation of ROS, 4-HNE and NOX4, expression of MMP-3, MMP-13, ADAMTS-4, ADAMTS-5 and degradation of the articular ECM in primary human chondrocytes. These findings suggest a novel role of the selective GLP-1 agonist lixisenatide in the treatment and prevention of OA. Further in vivo research using animal models or human trials is necessary to further validate these findings.
Disclosure statement
None of the authors of this study have any conflicts of interest that need to be disclosed.
Additional information
Funding
Related Research Data
References
- Cross M, Smith E, Hoy D, et al. The global burden of hip and knee osteoarthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73:1323–1330.
- Qu H, Li J, Wu L, et al. Trichostatin A increases the TIMP-1/MMP ratio to protect against osteoarthritis in an animal model of the disease. Mol Med Rep. 2016;14:2423–2430.
- Goldring MB, Otero M, Plumb DA, et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur Cell Mater. 2011;21:202.
- Nowotny K, Jung T, Höhn A, et al. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015;5:194–222.
- Ott C, Jacobs K, Haucke E, et al. Role of advanced glycation end products in cellular signaling. Redox Biology. 2014;2:411–429.
- Yu Z, Visse R, Inouye M, et al. Defining requirements for collagenase cleavage in collagen type III using a bacterial collagen system. J Biol Chem. 2012;287:22988–22997.
- Manka SW, Carafoli F, Visse R, et al. Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1. Proc Nat Acad Sci. 2012;109:12461–12466.
- Antonicelli F, Bellon G, Debelle L, et al. Elastin-elastases and inflamm-aging. Curr Top Dev Biol. 2007;79:99–155.
- Fields GB. Interstitial collagen catabolism. J Biol Chem. 2013;288:8785–8793.
- Gudmann NS, Karsdal MA. Type II Collagen. In: Karsdal MA, editor. Biochemistry of Collagens, Laminins and Elastin. Amsterdam, Nederland: Elsevier; 2016. p. 13–20.
- Ismail HM, Yamamoto K, Vincent TL, et al. Interleukin-1 acts via the JNK-2 signaling pathway to induce aggrecan degradation by human chondrocytes. Arthritis & Rheumatol. 2015;67:1826–1836.
- Ismail HM, Yamamoto K, Vincent T, et al. A significant role for the JNK pathway in regulating interleukin 1 induced-aggrecan degradation in human articular chondrocytes. Osteoarthritis and Cartilage. 2015;23:A152.
- Verma P, Dalal K. ADAMTS-4 and ADAMTS-5: key enzymes in osteoarthritis. J Cell Biochem. 2011;112:3507–3514.
- Stannus O, Jones G, Cicuttini F, et al. Circulating levels of IL-6 and TNF-α are associated with knee radiographic osteoarthritis and knee cartilage loss in older adults. Osteoarthr Cartil. 2010;18:1441–1447.
- Forbes JM, Cooper ME, Oldfield MD, et al. Role of advanced glycation end products in diabetic nephropathy. J Am Soc Nephrol. 2003;14:S254–S258.
- Roman-Blas JA, Jimenez SA. NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthr Cartil. 2006;14:839–848.
- Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373:2247–2257.
- McClean PL, Hölscher C. Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer’s disease. Neuropharmacology 2014;86:241–258.
- Vinué Á, Navarro J, Herrero-Cervera A, et al. The GLP-1 analogue lixisenatide decreases atherosclerosis in insulin-resistant mice by modulating macrophage phenotype. Diabetologia 2017;60:1801–1812.
- Lee YS, Jun HS. Anti-inflammatory effects of GLP-1-based therapies beyond glucose control. Mediators of Inflam. 2016;2016:1.
- Hansen MS, Tencerova M, Frølich J, et al. Effects of gastric inhibitory polypeptide, glucagon‐like peptide‐1 and glucagon‐like peptide‐1 receptor agonists on Bone Cell Metabolism. Basic Clin Pharmacol Toxicol. 2018;122:25–37.
- Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007;132:2131–2157.
- Berenbaum F, Bougault C, Attali C, inventors, Assistance Publique Hopitaux de Paris, assignee. Treatment of osteoarthritis with incretin hormones or analogues thereof. U.S. Patent No. 9,592,272. 2017 March 14.
- FDA. Sanofi new drug application for lixisenatide accepted for review by FDA; 2013 Feb 19; Available from: Drugs.com/PRNewsire.
- Kim Y, Zheng X, Ansari Z, et al. Mitochondrial aging defects emerge in directly reprogrammed human neurons due to their metabolic profile. Cell Rep. 2018;23:2550–2558.
- Smith MD, Triantafillou S, Parker A, et al. Synovial membrane inflammation and cytokine production in patients with early osteoarthritis. J Rheumatol. 1997;24:365–371.
- Fernandes JC, Martel‐Pelletier J, Pelletier JP. The role of cytokines in osteoarthritis pathophysiology. Biorheology 2002;39:237–246.
- Makki MS, Haqqi TM. Histone deacetylase inhibitor vorinostat (SAHA) suppresses IL-1β-induced matrix metallopeptidase-13 expression by inhibiting IL-6 in osteoarthritis chondrocyte. Am J Pathol. 2016;186:2701–2708.
- Heinrich PC, Behrmann I, Serge H, et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20.
- Lim H, Kim HP. Matrix metalloproteinase-13 expression in IL-1β-treated chondrocytes by activation of the p38 MAPK/c-Fos/AP-1 and JAK/STAT pathways. Arch Pharm Res. 2011;34:109–117.
- Mohetaer M, Li G, Wang Y, et al. Protective effects of gemigliptin against type II collagen degradation in human chondrocytes. Biomed Pharmacother. 2018;104:590–594.
- Mengshol JA, Vincenti MP, Coon CI, et al. Interleukin‐1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c‐jun N‐terminal kinase, and nuclear factor κB: differential regulation of collagenase 1 and collagenase 3. Arthritis & Rheumatol. 2000;43:801–811.
- Zandi E, Rothwarf DM, Delhase M, et al. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–252.