
Abstract
Bioavailability of the colloidal particles is of great concern. It is because about 40% of the newly discovered colloidal agents are poorly water soluble and thus poorly bioavailable. Various colloidal technologies have been adopted to enhance the bioavailability of such drugs. In this review, advanced colloidal technologies are discussed to understand the mechanism of bioavailability enhancement of drugs, coding latest references from the literature. Additionally, advantages and disadvantages of the concerned colloidal technologies are discussed to understand the potential usage of each process. All these colloidal technologies effectively enhance the bioavailability of poorly bioavailable drugs, either by protecting them from the environment of the gastrointestinal or by prolonging their intended therapeutic effect. Furthermore, complexation of the drug molecule with potential carries, particle size reduction to nanoscale, co-crystallization and self-emulsifying drug delivery system are also discussed which not only enhance the bioavailability but also reduce the potential toxicities of the loaded drugs. It is therefore important to understand the vital colloidal technologies with their recent advancements, potential applications and future challenges, which is discussed in this review. Finally, this article concludes that the advanced colloidal technologies, including freeze-dried liposome, solid lipid nanoparticle, polymeric nanoparticle (polymeric micelles), dendrimers and solid emulsifying drug delivery systems, have the potential to achieve improved bioavailability and targeted drug delivery.
PUBLIC INTEREST STATEMENT
To obtain the desired therapeutic effect of drugs, a preferred concentration of the drugs must reach the blood circulation. Most of the times, the desired concentration of drugs fails to reach the systemic circulation. This condition is also known as poor bioavailability of the drugs. This could be attributed to their poor water solubility, low absorption profile, first pass metabolism and degradation due to harsh gastrointestinal environment. A number of techniques are utilized to enhance the bioavailability of drugs. In this article, advance colloidal technologies are discussed, including freeze-dried liposomes, solid lipid nanoparticles, polymeric nanoparticles, dendrimers and solid emulsification with particular emphasis on their preparation, characterization and potential applications for the enhancement of bioavailability of drugs. These technologies not only improve the bioavailabilities of drugs but at the same time also result in targeted, site-specific and controlled drug delivery, thus ensuring no toxicities of the drug molecules.
1. Introduction
Bioavailability is the unchanged fraction of a drug in the blood circulation, which is available for its therapeutic use. It is one of the basic pharmacokinetic (PK) property of drugs. According to a definition, an intravenously administered drug has 100% bioavailability (Griffin & O’Grady, Citation2006). It also refers to the rate at which the active pharmaceutical ingredient (API) enters systemic circulation, reaching the site of action. Solubility of a drug in water is the basic property which leads to the absorption of the drug after oral intake. It is also useful in accessing, manipulating and testing of drug properties during its developmental design and procedure (Williams et al., Citation2013). However, the bioavailability of oral drugs depends upon multiple factors including their water solubility, drug permeability across the gastrointestinal (GIT) membrane, the rate of dissolution in GIT fluid, first-pass effect of the liver enzymes on drug metabolism and susceptibility to the efflux mechanisms (Krishnaiah, Citation2012). The number of insoluble drug candidates in drug discovery has increased in recent years, as about 70% of the new drug candidates have shown poor water solubility (Kawabata, Wada, Nakatani, Yamada, & Onoue, Citation2011). So, for these drug candidates, the limiting factor in in-vivo bioavailability of oral drugs is their poor water solubility, dissolution and disintegration in the GIT fluids. Therefore, in vitro dissolution study has been acknowledged as prominent factor in new drug development. Thus, enhancing the bioavailability of poorly bioavailable drugs by increasing their dissolution rate is a main challenge for pharmaceutical scientists of modern age (Hu, Johnston, & Williams, Citation2004; Wakaskar, Citation2017a).
In earlier decades, the foremost objectives of product development were to increase bioavailability, stability and degradation of the drugs which ultimately leads to the patient compliance. The bioavailability of an oral drug was chiefly influenced by its water solubility at certain pH (1.0–7.5) and its frequency of transmission through the biological membranes (Vemavarapu, Mollan, Lodaya, & Needham, Citation2005). Inadequate dissolution and disintegration rate were the primary limiting factors in the bioavailability of poorly aqueous soluble oral drugs. In this respect, various novel methods and innovative technologies have currently been introduced for the prime purpose of enhancing wettability and water solubility of (active pharmaceuticals agents) APIs (Komal et al., Citation2017; Maria et al., Citation2017). The different methods, being commonly used for this purpose, were categorized as physical and chemical reforms. The physical reform includes reduction of particle size of API to enhance the surface area (micronization and nanonization), formation of polymorphs/pseudopolymorphs which includes solvates, complexation/solubilization (by means of using surfactants or cyclodextrins [CDs]), conjugating to dendrimers, by addition of cosolvents for increasing solubility and preparation of drug dispersions in carriers (eutectic mixtures, non-molecular solid dispersions and solid solutions). Similarly, various chemical reforms are done to enhance the bioavailability of drugs including the synthesis of soluble prodrugs and salts (Chen, Khemtong, Yang, Chang, & Gao, Citation2011; Gao, Zhang, & Chen, Citation2008).
In this review, various advanced colloidal technologies and approaches have been discussed which are effectively used for the enhancement of bioavailability of poorly water-soluble rugs. Some of them include the nano-particulate technology, polymeric micelles (PMs), solid emulsifying drug delivery system (SEDDS) and complexation with beta (β)-CD. A representative diagram of all the techniques is given in Figure . Moreover, some formulation-based approaches are also discussed here.
Figure 1. Scheme of strategies used for enhancing the bioavailability of poorly water-soluble drugs.
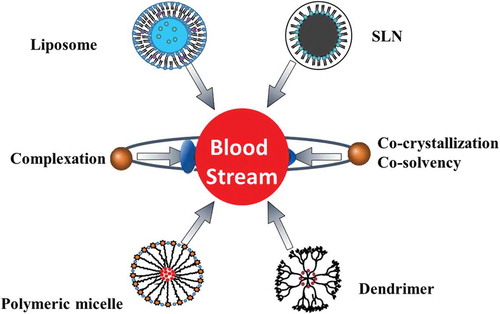
2. Particle technologies
Particle technology is a bunch of techniques, used to enhance physicochemical, micrometrics and biopharmaceutical characteristics of the hydrophobic drugs, resulting in their improved solubility and bioavailability. Such techniques include reduction of the particle size, modification of the crystal development habit and use of polymers and copolymers (Salman et al., Citation2016; Savjani, Gajjar, & Savjani, Citation2012). Similarly, some of these nanosized particle technologies include freeze-dried liposomes, solid lipid nanoparticle (SLN) and dendrimers.
2.1. Freeze-dried liposomes
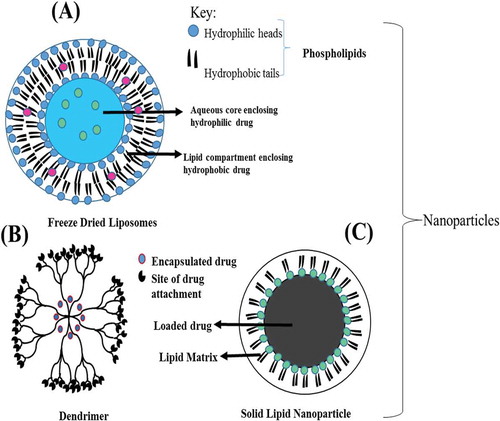
Liposomes are the phospholipid vesicles, comprising entirely of a phospholipid bilayer which surrounds an aqueous compartment within its hydrophilic head and thus can dissolve both lipophilic and hydrophilic drugs. Lipophilic drugs are entrapped in their lipid domain while hydrophilic drugs are taken up in their aqueous core (Figure ) (Fahr & Liu, Citation2007). Because of their dual biphasic characteristics, variety in design and composition, they propose a great dynamic technology for improving the drug solubility of poorly water-soluble drugs (Alam et al., Citation2016). Drugs loading into liposomes leads to significant improvements in their PK and pharmacodynamic properties as compared to their free forms, resulting in increased therapeutic effect and decreased toxicity of the loaded drugs (Zeb et al., Citation2017; Zhang et al., Citation2005). Nevertheless, one of the major setback with application of liposomes as drug delivery systems is their poor stability on storage. The liposomal formulations can therefore be stabilized by freeze drying the liposomes to get dry powders with increased stability and uphold potency of the incorporated drug. Ghanbarzadeh (Citation2013) found that freeze-dried liposomes of sirolimus (rapamycin) have increased stability after reconstitution in association with the conventional suspension product of the same drug (Ghanbarzadeh, Valizadeh, & Zakeri-Milani, Citation2013). Moreover, he observed that the stability of the formulation was extraordinarily improved when dextrose was used as a lyoprotectant during freeze drying. He concluded that freeze-drying technique may improve the stability of liposomes. Furthermore, in the production of paclitaxel-liposomal formulation, it was observed that numerous sugars can be used as lyoprotectants including sucrose, dextrose and trehalose (Zhang et al., Citation2005). This formulation was sterile and easy to handle with increased solubility as well as enhanced physicochemical stability. In another study, paclitaxel-loaded liposomes were developed with polyethylene glycol (PEG) 400 in the hydration medium of liposome resulting in improved solubilization and drug entrapment, leading to improved bioavailability of paclitaxel in rats (Yang et al., Citation2007). Few representative studies consuming liposomes as efficient drug delivery system with enhanced therapeutic effects of the loaded drugs include augmented inhalation therapy using insulin (Bi, Shao, Wang, & Zhang, Citation2008) and oseltamivir phosphate (Tang et al., Citation2015) and antioxidant properties of curcumin (Takahashi, Uechi, Takara, Asikin, & Wada, Citation2009). Freeze-dried liposome system, being a favorable approach for developing liposomal formulation with enhanced water solubility and upheld bioavailability, could therefore be used for a variety of poorly water-soluble therapeutic agents.
Figure 2. Nanoparticle: (A) freeze-dried liposome, (B) dendrimer, (C) SLN.
2.2. SLNs
SLNs are colloidal drug carrier system that are developed using lipid which are solid at room temperature. Solid lipid includes glycerides, waxes having high melting points (Üner & Yener, Citation2007). SLNs got recent attention because of their ability to replace traditional, orthodox colloidal carrier systems such as emulsions and suspensions (Muèller, Maèder, & Gohla, Citation2000; ud Din, Mustapha et al., Citation2015). Various methods are reported for SLN preparation including hot and cold homogenization high pressure homogenization (HPH), degradation of o/w emulsion droplets, solvent injection, w/o/w double emulsion, high shear, stress homogenization (mixing) and dispersion using ultrasonic waves. Among these, HPH is the most efficient method owing to the formation of narrow particles with uniform sizes, greater particle content and organic solvents avoidance (ud Din, Mustapha et al., Citation2015; Üner & Yener, Citation2007). Moreover, the SLN is preferred over traditional colloidal carrier system mainly due to its controlled drug release, targeted drug delivery, enhanced stability, bio-friendly nature of the carrier and the capacity to incorporate both hydrophilic and lipophilic drugs (Figure ). Besides their numerous advantages, SLN exhibited several disadvantages including low drug loading capacity and stability issues during storage or drug administration. Various studies have been performed to check the effectiveness of SLN in enhancing the solubility of poorly soluble aqueous drug (ud Din, Rashid et al., Citation2015). One such study was performed using ATRA (all-trans-retinoic acid) for oral administration by incorporating the drug in SLN particles. It was observed that the absorption of ATRA was significantly enhanced, resulting in improved bioavailability of the drug. In another study, a hydrophobic drug, tretinoin, was incorporated into SLN by using emulsification method. It demonstrated a sustained release of the drug from SLN with improved stability (Das, Ng, Kanaujia, Kim, & Tan, Citation2011). Similarly, uh Din, Aman et al. (Citation2017) exhibited that the SLN incorporated drug not only improves the bioavailability of the antitumor drugs but also reduces the side effects of potentially cytotoxic drug, irinotecan (ud Din, Cho et al., Citation2017). Moreover, they demonstrated that SLN, if further entrapped in hydrogel, may result in excellent sustained release of the loaded drug. Also, this system was observed to be stable for 6 months, which was only possible because of the exceptional entrapment efficiency of SLN drug delivery system. Thus, these studies revealed the possibility of using SLN drug delivery system for controlled and prolonged drug delivery. Some other examples of SLN as drug delivery systems for enhanced bioavailability of the colloidal drugs include oral bioavailability of curcumin (Kakkar, Singh, Singla, & Kaur, Citation2011), enhanced GIT availability of quercetin (Li et al., Citation2009) and irinotecan (uh Din, Mustapha, & Kim, Citation2017).
2.3. Dendrimers
These are the macromolecules, globular in structure, highly branched, with many arms originating from a core (Tomalia & Frechet, Citation2002). Moreover, they proved to be novel approach to increase the bioavailability of poorly aqueous soluble drugs. Dendrimers are produced by a stepwise process that includes a molecule with highly regular branched pattern. Furthermore, dendrimer possess a distinct, entirely unique molecular weight, low polydispersity index and a well-defined number of groups on the periphery as can be seen in Figure . Dendrimers were first reported about two decades ago, but those studies were only concerned with their synthesis, physical and chemical nature. It was only recently, when their studies become more therapeutic oriented. Dendrimers have shown great results in various fields ranging from genetics (gene delivery) to magnetic resonance imaging, to the production of vaccines, antiviral, antibacterial and anticancer drugs (Talanov et al., Citation2006).
A variety of applications of dendrimers have been found in the field of pharmaceutical and medical chemistry, a comprehensive information of which can be found in a recent review covering the area of dendrimers (uh Din, Aman et al., Citation2017). Dendrimers, as drug delivery systems, were used to entrap the drug molecules onto the surface of dendrimers or other functionalities. The reason why the dendrimers have become the excellent candidates as drug carriers is because of their proper, well-defined structure, compact size, globular shape, mono-dispersity size and controlled “surface” functionalities. Despite the potential application of dendrimers, little studies have actually been done in this area. Following are some of the few illustrative studies using colloidal drugs including indomethacin (Chauhan et al., Citation2003), camptothecin (Jain & Gupta, Citation2008) and doxorubicin (Ke, Zhao, Huang, Jiang, & Pei, Citation2008).
Chauhan et al. reported the transdermal delivery of aqueous formulations of indomethacin using dendrimers. They observed that the dendrimers significantly enhanced the steady state flux of indomethacin, more exactly 4.5 times, as compared to its pure form. Moreover, similar results were revealed in in vivo studies which demonstrated around twofold bioavailability as compared to pure drug (Chauhan et al., Citation2003). Furthermore, NK Jain and Guptha reported the applications of dendrimer–drug complexation to achieve the augmented drug solubility and bioavailability. More particularly, the campthothecin, an anticancer drug bioavailability, was significantly enhanced using dendrimer-based drug delivery system (Jain & Gupta, Citation2008). Similarly, Ke et al. (Citation2008) reported the development of doxorubicin loaded-PAMAM dendrimers for oral administration. They observed that dendrimer complex resulted in enhanced cellular uptake and 4–7 times more transport capability of the loaded drug as compared to the free doxorubicin. They concluded that doxorubicin-loaded dendrimer may enhance the bioavailability of the loaded drug by more than 200-fold after oral administration as compared to the pure drug.
3. PMs
PMs are nanosized formulations which have the capacity to carry water insoluble drugs to their targeted areas for their intended pharmacological effects. They are smaller in size (100 nm) and can swallow up infected cells by system of macrophages (Jones & Leroux, Citation1999; Wakaskar, Citation2017b, Citation2017c). Being amphiphilic in nature, the di- or tri-block copolymers thus contain both hydrophobic and hydrophilic segments. These amphiphilic block copolymers when exposed to aqueous environment, above certain concentration (called critical micelle concentration or CMC), form micelles. The hydrophobic segment of block copolymer constitutes the core of micelle, while hydrophilic segment forms the shell of micelles Figure . Due to specific core-shell structure, hydrophobic drugs are carried by hydrophobic core and the hydrophilic shell stabilize water-soluble drugs (Otsuka, Nagasaki, & Kataoka, Citation2003). This ensures the PM’s solubility in aqueous medium and also controls their in vivo PK (Gothwal, Khan, & Gupta, Citation2016; Wakaskar, Citation2018a). Some commonly used methods for the preparation of PMs are the dialysis method, oil-in-water emulsion method, solvent evaporation method, cosolvent evaporation method and freeze-drying method (Danhier, Feron, & Preat, Citation2010; Gaucher et al., Citation2005; Rapoport, Citation2007). PEG corona increases the strength of PMs so that they can circulate in blood more efficiently and target the solid tumor directly through enhanced permeability and retention (EPR) effect (Danhier et al., Citation2010; uh Din et al., Citation2017; Wakaskar, Citation2018b). EPR effect is produced due the accumulation of PMs at the solid tumor location, which have poor lymphatic drainage system leading to insufficient vascular retrieval of the extravasated molecules. Micelle’s surface has anticancer antibody which shows interaction with certain receptors on cancerous cells. Vascularization of tumors releases specific chemicals which makes micelle accumulation possible in it. Principally, PMs are designed to release drug slower in to the blood stream so that they can efficiently kill tumorous cells (Rapoport, Citation2007; Sun & Zeng, Citation2002). One such study was performed by encapsulating doxorubicin in PMs, which releases its encapsulated drug by varying pH. Doxorubicin targets the cancerous cell’s nucleus and accumulates in it, after reaching there, it enters into its DNA and interacts with DNA polymerase enzyme. As a result DNA, is cleaved and cell death occur (Sun & Zeng, Citation2002). In an-other study anticancer drug paclitaxel and anti-fungal drug amphotericin B were allowed to solubilize into hydrotropic PMs by dialysis method. As a result their stability and biological availability were enhanced (Lee et al., Citation2007). Some examples of poorly water-soluble anticancer drugs delivered through PMs include paclitaxel, etoposide, docetaxel and 17-allylamino-17-demethyoxygeldanamycin. All these drugs bioavailability were enhanced through this system (Shin, Alani, Rao, Rockich, & Kwon, Citation2009).
Figure 3. Schematic of polymeric nanomicelle.
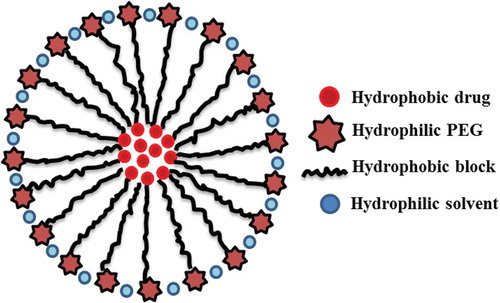
Similarly, oral drugs have certain limitations regarding solubility due to harsh environment of GI tract. Presence of digestive enzymes/bile salts, variation in pH causes the destruction of orally administered drugs. To avoid such degradation, thermodynamically and kinetically stable PMs were prepared (Calderara et al., Citation1994). Thermodynamic stability was achieved when copolymer concentration was above its CMC. Kinetic stability was achieved even when copolymer concentration was below its CMC. Thus required amount of drug reached the target site resulting in intact oral availability (Adams, Lavasanifar, & Kwon, Citation2003). Various PMs were developed to enhance the bioavailability of poorly water-soluble drugs, including pH-sensitive PMs which were used to promote the controlled release properties of loaded drugs at target region, muco-adhesive PMs for prolonging the residence time in the gut and P-glycoprotein inhibitors PMs for prolonging the drug accumulation (Xu, Ling, & Zhang, Citation2013).
4. SEDDS
According to biopharmaceutical classification system, drugs that belong to class 2, 3 and 4 can be used for the development of SEDDS, However, class 2 drugs are more specifically used (Gupta, Kesarla, & Omri, Citation2013). SEDDS are mixture of surfactant, oil and cosolvent which form isotropous formulation; these formulations emulsify freely in an aqueous phase with mild agitation to form oil in water emulsions (Bhatt, Citation2004). The digestive movement of stomach and small intestine leads to their slight agitation (Shah, Carvajal, Patel, Infeld, & Malick, Citation1994). The emulsion formed in SEDDS has a particle size of 100–300 nm. An advanced form of this technique is recently recognized, known as self-micro emulsification, in which the particle size is less than 50 nm (Gursoy & Benita, Citation2004). Reduction to submicron level of the particle size leads to increased surface area for absorption eradicating the dissolution step. It also results in prolongation of the drug absorption leading to increased bioavailability (Gupta et al., Citation2013) and decreased chemical and enzymatic deterioration in the aqueous environment of GIT.
SEDDS is evolved by the integration of the liquid or semi-solid emulsifying ingredients into powders or nanoparticles by various methods such as spray drying (Chauhan, Shimpi, & Paradkar, Citation2005), spray cooling (Rodriguez et al., Citation1999), melt granulation (Seo & Schæfer, Citation2001) and adsorption to solid carriers leading to the formation of solid dispersion (Rashid, Din et al., Citation2015; Rashid, Kim et al., Citation2015) that can be converted into other solid dosage forms like pellets and tablets microsphere, suppositories and implants. Another form is liquid SEDDS which is formed as liquid dosage form or liquid filled soft gelatin capsules, but they have some constraints including their high cost and difficult intake, and storage problems. Therefore, solid SEDDS are preferred over the liquid SEDDS system (Kim, Kang, Oh, Yong, & Choi, Citation2012). Excipients are chosen by dissolving the drug in a mixture of oil surfactant and cosolvent. Many variables are considered while formulating the SEDDS system such as the droplet size, surfactant and concentration of surfactant and surfactant/oil ratio which determine the self-emulsifying nature of the formulation as can be seen in Figure . Thus, only few combinations are acceptable to form an SEDDS system (Patel, Kevin, Patel, Raval, & Sheth, Citation2011).
Figure 4. Development of self-emulsifying drug delivery system.
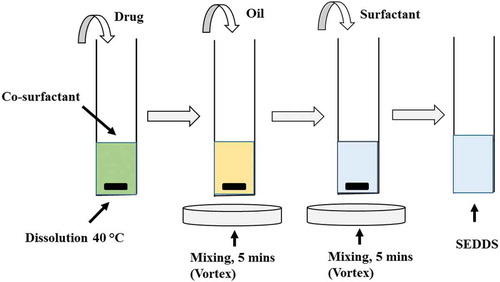
One of the main ingredients of SEDDS are oils as they enhance the solubilization of lipophilic drugs leading to increased bioavailability. Triglycerides in large and medium size are most widely used (Khasia & Khasia, Citation2012). Similarly, most desirable among the surfactants are the nonionic surfactants as they have high hydrophilic lipophilic balance values; they enhance the O/W droplet formation resulting in quick dispersion of the formulation in the aqueous media. Typically used surfactant strength limits from 30% to 60% and excessive amount of surfactant can result in GIT irritation (Ayushi, Faraz, Ritika, & Saurabh, Citation2012). However, in SMEDDS, insignificantly higher amount of surfactant is used (40–60%) (Grove, Müllertz, Nielsen, & Pedersen, Citation2006). Furthermore, cosolvents like propylene glycol, PGE and polyoxyethylene assist in dissolving large amount of hydrophilic surfactant or hydrophobic drug in lipid base. These cosolvents lower the interfacial tension to a temporary negative value, leading to production of fine droplets which absorb more surfactant and cosurfactant, till their bulk condition adequately exhausted to reobtain a positive value of interfacial tension (Nigade, Patil, & Tiwari, Citation2012).
A commercially available SEDDS cyclosporine, resulted in twofold bioavailability in humans when compared with other cyclosporine formulations (Mueller et al., Citation1994). Moreover, SNEDDS is recently being introduced for flutamide (an antiandrogen drug for the treatment of prostate cancer) and telmisartan which is an angiotensin II receptor blocker antihypertensive agent (Patel et al., Citation2011). Other examples of the drugs prepared in the form of solid self-emulsifying system are nimodipine (Kim et al., Citation2012), flurbiprofen (Seo & Schæfer, Citation2001) docetaxel (Chen et al., Citation2011), curcumin (Yan et al., Citation2011), meloxicam (Agarwal, Alayoubi, Siddiqui, & Nazzal, Citation2013) and fenofibrate (Kanaujia, Ng, & Tan, Citation2014).
5. Complexation with β-CD
CDs are cyclic oligosaccharides made up of an adjustable number of glucose units, interlinked by 1,4 bonds. When starch counters with CD-glycosyltransferase enzyme (CGTase), CDs are formed (Thompson, Citation1997). Representative CDs comprise 6–8 units of glucose monomers in a ring, generating a cone shape: α (alpha)-CD: (6-membered), β-CD: (7-membered) and γ (gamma)-CD: 8-membered sugar ring molecule. α- and γ-CD are mostly used in the nutrition industry. The CD molecules are hydrophilic superficially and hydrophobic inside their ring cavity. In liquid or sometimes solid media, inclusion complexes are designed by CD with diverse type of erratically sized, nonpolar molecules (Frömming & Szejtli, Citation1994). In compounds with lower solubility, inclusion in CDs enhance their solubility and dissolution. With intermediate complex stability constant values, the active ingredient easily enters the membranes leading to increased bioavailability and therapeutic effectiveness (Loftsson, Sigurđsson, Másson, & Schipper, Citation2004). It is stated that organic acids (especially citric acid) and CDs have synergistic influence on the solubility of drugs, which could be attributed to the adaptation in solute–solvent interrelationship. Solubility of compounds depends on the type of CDs used for their preparation. Improved solubility is achieved when derivatized CDs are used in place of non-derivatized CDs owing to the highly soluble nature of derivative CDs. Derivatization improves aqueous solubility, as results in hydroxypropyl-β-CD, by conveying better flexibility to the exterior CD hydroxyl groups. Moreover, it has the capability to upsurge hydrogen bond formation between the guest molecule and the derivatized CD leading to the formation of more stable inclusion complex. Tomasik and Schilling (Citation1998) optimized several CDs compositions of ABZ inclusion complex and found that CD, 2-hydroxypropyl-β-CD appeared to be more suitable grounded on completion behavior and safety for human beings.
It was observed that among the various CDs, β-CD is most appropriate for the manufacture of pharmaceutical preparations. It could be attributed to its low-cost production and easy method of separation from mixture (Del Valle, Citation2004). Naproxen (Mura, Faucci, & Bettinetti, Citation2001), piroxicam (Sauceau, Rodier, & Fages, Citation2008), ketoconazole (KET) (Taneri, Ozcan, & Guneri, Citation2010), ibuprofen (Ghorab & Adeyeye, Citation2003), ketoprofen (Ann, Kim, Choi, & Kim, Citation1997) and ABZ (Tomasik & Schilling, Citation1998) are the most frequent drugs with complex formation in order to improve their bioavailability (Table ).
Table 1. Bioavailability enhancement through complexation
One of the most common examples of β-CD used in augmenting solubility of drugs is albendazole (ABZ) complexation with β-CD. ABZ, an anthelminthic drug, has been effectively employed against human and animal helminthic parasites, such as nematodes (Saenger, Citation1980). However, its very poor solubility profile leads to a truncated absorbance though the GIT leads to GI turbulences and severe side effects (Pitha, Milecki, Fales, Pannell, & Uekama, Citation1986). Another significant example is the use of hydroxyl propyl-CD (extremely soluble CD derivative) for increasing the solubility, oral bioavailability and dissolution rate of very poorly water-soluble anti-fungal agent KET. On evaluation by HPLC method, tablets containing cyclodexttrin complex show higher bioavailability and dissolution rate as compared to the KET alone (Taneri et al., Citation2010). The solubility of naproxen was also increased by combined effect of hydroxypropyl-β-CD complexation and polyvinylpyrrolidone (Mura et al., Citation2001).
6. Formulation-based approaches
Out of all newly discovered compound known in medication disclosure projects, 470 compounds are inadequately soluble in water (Allam, El. Gamal, & Naggar, Citation2011). The increase in the extent of poorly soluble compounds can be accredited to improvements in synthesis technology, which have permitted the planning of dreadfully sophisticated compounds, and an alteration in discovery approach from a supposed configuration methodology to a targeted methodology (Kawakami, Citation2012). Poorly bioavailable drugs are a great challenge for the formulation scientists to develop suitable dose forms with enhanced bioavailability and effective therapeutics. To address this challenge, poorly bioavailable drugs are mostly prepared in three different forms which includes crystalline solid formulations, amorphous formulations and lipid formulations. The formulation-based approaches used for this purpose include co-crystallization (Shiraki, Takata, Takano, Hayashi, & Terada, Citation2008), cosolvency (Strickley, Citation2000) and hydrotrophy (Badwan, El-Khordagui, Saleh, & Khalil, Citation1982).
6.1. Co-crystallization
Co-crystals mainly comprise two parts; the API and the former. Currently, the former is any other excipient or API that once given together reduces the dose amount and side effects. Co-crystallization is an effective crystal engineering method which results in diverse properties of the drugs by altering their crystal structure. An extra refined meaning of a co-crystal is “multicomponent crystal that is molded between two compounds that are solids under ambient conditions, wherever at least one component is an appropriate ion or molecule”. Some drugs advertised within the style of racemic co-crystals include atenolol, atropine, certirazine, fluoxetine, ketoprofen, loratadine, omeprazole and zopiclone. Pharmaceutical co-crystals are non-ionic supramolecular networks that can be employed to enhance the physical properties of pharmaceuticals without altering the chemical preparations of the API (Miroshnyk, Mirza, & Sandler, Citation2009).
This complex is formed by numerous styles of interaction, as well as pi-stacking, hydrogen bonding and van der Waals forces. In nonionizable compounds, co-crystals improve pharmaceutical properties by adjustment of chemical stability, mechanical behavior, solubility, dissolution rate and bioavailability (Shiraki et al., Citation2008). Approaches used for enhancing the bioavailability of poorly water-soluble drugs using co-crystallization technique include solvent evaporation method (Mutalik, Anju, Manoj, & Usha, Citation2008), grinding (Sekhon, Citation2005), slurring (Ober & Gupta, Citation2012), supercritical fluid atomization (Breitenbach, Citation2002), hot melt extrusion (Miller, McConville, Yang, Williams, & McGinity, Citation2007) and sono-crytalization (Bucar & MacGillivray, Citation2007).
In future, co-crystallization analysis can significantly improve the development of newest synthons, which are the structural entities for developing a crystal. As soon as the flexibility in crystal structure style and prediction progresses, the high magnitude approach are progressively marginalized. Conversely, the diverse crystallization conditions delivered by high-throughput screening experiments usually result in electrifying innovations. Some of the representative studies using co-crystal approaches include co-crystallization of pterostilbene: Caffeine (Mutalik et al., Citation2008) and co-crystals of Cox-2 inhibitor & amp, venlafaxine (Sekhon, Citation2005).
6.2. Cosolvency
The solubility of a poorly water-soluble drug is improved normally by adding a water-soluble solvent, these solvents are called cosolvents (Strickley, Citation2004). Cosolvents are mixtures of water and one or more water-soluble solvents employed to obtain a solution with amplified solubility. Conventionally, this is one of the mostly used techniques for increasing the bioavailability of poorly water-soluble drugs. Examples of solvents employed in cosolvent mixtures are PEG 300, propylene glycol or ethanol. Cosolvent preparations of poorly soluble drugs are administered orally and parentally. Parenteral preparations usually require the addition of water or a dilution step with a liquid media to decrease the solvent concentration before administration. Cosolvents upsurge the solubility of poorly soluble compounds thousand-times in association with the solubility of drugs in their pure form.
Cosolvents in combination with different solubilization methods and pH adjustment are highly recommended for enhancing the bioavailability of poorly bioavailable drugs (Millard, Alvarez-Nunez, & Yalkowsky, Citation2002). Dimethylsulfoxide and dimethylacetoamide are extensively used cosolvents for this purpose, owing to their great solubilization capability and comparatively reduced toxicity. Co-dissolvable items, Nimodipine infusion (Nimotop®, Bayer) and digoxin Elixir pediatric (Lanoxin®, GSK), are examples of co-dissolvable formulations (Seedher & Bhatia, Citation2003).
6.3. Hydrotrophy
Hydrotrophy was introduced by Carl neuberg in 1916, however, practically it was implicated by thoma and colleagues in 1976 (Sekhon, Citation2005). In this approach a large amount of secondary substance is added to improve the aqueous solubility of poorly water drugs. Hydrotropes comprise each associated degree, anionic cluster and a hydrophobic aromatic ring or ring system (Table ). The anionic cluster enhances the hydrophilicity; thus, the ring system cooperates with the substance to be dissolved (Nidhi, Indrajeet, Khushboo, Gauri, & Sen, Citation2011). The mechanism involved in hydrotropy is associated to complex formation that comprises interaction between lipophilic drug and therefore the hydrotropic agents like urea, nicotinamide, sodium alginate and sodium benzoate (Zaheer, Naveen, Santosh, & Imran, Citation2011) Hydrotropy is extensively used in the formulation of dry syrups of poorly soluble drugs. Measurable assessments of poorly water-soluble drugs by UV–visible spectrophotometric analysis prevent the consumption of organic solvents and quantifiable assessments of poorly water-soluble drugs by titrimetric analysis, such as ibuprofen and flurbiprofen.
Table 2. Categorization of hydrotropes
7. Conclusion
Bioavailability in a specific range is required for all therapeutic agents in order to achieve their intended effects. The bioavailability can be achieved by directing the drugs to their specific site of action, reducing the particle size, loading it into a potential drug carrier system and protecting them from the tough environment of the GIT. Moreover, prolonging their release over intended period of time and complexation with potential carries also enhance their bioavailability, resulting in improved therapeutic effects. All these goals of enhancing the bioavailability of the poorly water-soluble drugs can be achieved by using the advanced colloidal technologies.
Competing interests
The authors declare no competing interests.
Additional information
Funding
Notes on contributors

Fakhar ud Din
Dr. Fakhar-ud-Din is an assistant professor in Department of Pharmacy Quaid-i-Azam University Islamabad, Pakistan. He earned his PhD degree in Pharmaceutics from Hanyang University, South Korea. His research interest is multi-disciplinary including novel drug delivery systems, nanoparticles, colloidal technologies, anticancer drugs for therapeutic applications, nano-gels, anti-leishmanial drugs formulations and intervention-based pharmacy practice at national and community level. He has published >50 research papers in journals of international repute/international conference, including 3 international patents to his name so far. Most of the coauthors of this article are final year Doctor of Pharmacy students who have great interest in the field of drug delivery and nanotechnology to obtain the desired therapeutic effect of drugs with minimized toxicities. This article is one of the many such efforts from this research group, highlighting the importance of colloidal technologies to achieve maximum therapeutic effect of the existing and newly developed drug molecules. Moreover, these technologies reduces the side effects of the drugs.
Related Research Data
References
- Adams, M. L., Lavasanifar, A., & Kwon, G. S. (2003). Amphiphilic block copolymers for drug delivery. Journal of Pharmaceutical Sciences, 92(7), 1343–1355. doi:10.1002/jps.10397
- Agarwal, V., Alayoubi, A., Siddiqui, A., & Nazzal, S. (2013). Powdered self-emulsified lipid formulations of meloxicam as solid dosage forms for oral administration. Drug Development and Industrial Pharmacy, 39(11), 1681–1689. doi:10.3109/03639045.2012.729594
- Alam, Z., Salman, Q. O., Seo, K. H., Hye, C. J., Seong, K. H., & Ki, K. J. (2016). International Journal of Nanomedicine, 11, 3813–3824. doi:10.2147/IJN.S109565
- Allam, A. N., El. Gamal, S., & Naggar, V. (2011). Bioavailability: A pharmaceutical review. International Journal Novel Drug Delivery Technical, 1(1), 77–93.
- Ann, H. J., Kim, K. M., Choi, J.-S., & Kim, C. K. (1997). Effects of cyclodextrin derivatives on bioavailability of Ketoprofen. Drug Development and Industrial Pharmacy, 23(4), 397–401. doi:10.3109/03639049709146143
- Ayushi, T., Faraz, J., Ritika, S., & Saurabh, S. (2012). International Research Journal of Pharmacy, 3(5), 32-36.
- Badwan, A., El-Khordagui, L., Saleh, A., & Khalil, S. (1982). The solubility of benzodiazepines in sodium salicylate solution and a proposed mechanism for hydrotropic solubilization. International Journal of Pharmaceutics, 13(1), 67–74. doi:10.1016/0378-5173(82)90143-0
- Bhatt, P. P. (2004). The Drug Delivery Companies Report Autumn/Winter. Pharma Ventures Ltd, 1, 26.
- Bi, R., Shao, W., Wang, Q., & Zhang, N. (2008). Spray-freeze-dried dry powder inhalation of insulin-loaded liposomes for enhanced pulmonary delivery. Journal of Drug Targeting, 16(9), 639–648. doi:10.1080/10611860802201134
- Breitenbach, J. (2002). Melt extrusion: From process to drug delivery technology. European Journal of Pharmaceutics and Biopharmaceutics, 54(2), 107–117. doi:10.1016/S0939-6411(02)00061-9
- Bucar, D.-K., & MacGillivray, L. R. (2007). Preparation and reactivity of nanocrystalline cocrystals formed via sonocrystallization. Journal of the American Chemical Society, 129(1), 32–33. doi:10.1021/ja0671161
- Calderara, F., Hruska, Z., Hurtrez, G., Lerch, J., Nugay, T., & Riess, G. (1994). Macromolecules, 27(5), 1210. doi:10.1021/ma00083a020
- Chauhan, A. S., Sridevi, S., Chalasani, K. B., Jain, A. K., Jain, S. K., Jain, N., & Diwan, P. V. (2003). Dendrimer-mediated transdermal delivery: Enhanced bioavailability of indomethacin. Journal of Controlled Release, 90(3), 335–343. doi:10.1016/S0168-3659(03)00200-1
- Chauhan, B., Shimpi, S., & Paradkar, A. (2005). Preparation and evaluation of glibenclamide-polyglycolized glycerides solid dispersions with silicon dioxide by spray drying technique. European Journal of Pharmaceutical Sciences, 26(2), 219–230. doi:10.1016/j.ejps.2005.06.005
- Chen, H., Khemtong, C., Yang, X., Chang, X., & Gao, J. (2011). Nanonization strategies for poorly water-soluble drugs. Drug Discovery Today, 16(7), 354–360. doi:10.1016/j.drudis.2010.02.009
- Chen, Y., Chen, C., Zheng, J., Chen, Z., Shi, Q., & Liu, H. (2011). Development of a solid supersaturatable self-emulsifying drug delivery system of docetaxel with improved dissolution and bioavailability. Biological and Pharmaceutical Bulletin, 34(2), 278. doi:10.1248/bpb.34.278
- Danhier, F., Feron, O., & Preat, V. J. (2010). To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. Journal of Control Release, 148(2), 135–146. doi:10.1016/j.jconrel.2010.08.027
- Das, S., Ng, W. K., Kanaujia, P., Kim, S., & Tan, R. B. (2011). Formulation design, preparation and physicochemical characterizations of solid lipid nanoparticles containing a hydrophobic drug: Effects of process variables. Colloids and Surfaces B: Biointerfaces, 88(1), 483–489. doi:10.1016/j.colsurfb.2011.07.036
- Del Valle, E. M. (2004). Cyclodextrins and their uses: A review. Process Biochemistry, 39(9), 1033–1046. doi:10.1016/S0032-9592(03)00258-9
- Fahr, A., & Liu, X. (2007). Drug delivery strategies for poorly water-soluble drugs. Expert Opinion on Drug Delivery, 4(4), 403–416. doi:10.1517/17425247.4.4.403
- Frömming, K., & Szejtli, J. (1994). Cyclodextrins in pharmacy. Dordrecht: Kluwer Academic Publishers.
- Gao, L., Zhang, D., & Chen, M. (2008). Drug nanocrystals for the formulation of poorly soluble drugs and its application as a potential drug delivery system. Journal of Nanoparticle Research, 10(5), 845–862. doi:10.1007/s11051-008-9357-4
- Gaucher, G., Dufresne, M.-H., Sant, V. P., Kang, N., Maysinger, D., & Leroux, J.-C. (2005). Block copolymer micelles: Preparation, characterization and application in drug delivery. Journal of Controlled Release, 109(1), 169–188. doi:10.1016/j.jconrel.2005.09.034
- Ghanbarzadeh, S., Valizadeh, H., & Zakeri-Milani, P. (2013). Advanced Pharmaceutical Bulletin, 3(1), 25.
- Ghorab, M. K., & Adeyeye, M. C. (2003). Enhanced bioavailability of process-induced fast-dissolving ibuprofen cogranulated with β-cyclodextrin. Journal of Pharmaceutical Sciences, 92(8), 1690–1697. doi:10.1002/jps.10443
- Gothwal, A., Khan, I., & Gupta, U. (2016). Polymeric Micelles: Recent advancements in the delivery of anticancer drugs. Pharmaceutical Research, 33(1), 18–39. doi:10.1007/s11095-015-1784-1
- Griffin, J. P., & O’Grady, J. (2006). The textbook of pharmaceutical medicine. Wiley Online Library, Blackwell Publishing Ltd. London. doi:10.1002/9780470987391.
- Grove, M., Müllertz, A., Nielsen, J. L., & Pedersen, G. P. (2006). Bioavailability of seocalcitol. European Journal of Pharmaceutical Sciences, 28(3), 233–242. doi:10.1016/j.ejps.2006.02.005
- Gupta, S., Kesarla, R., & Omri, A. (2013). Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. ISRN Pharmaceutics, 2013. doi:10.1155/2013/848043
- Gursoy, R. N., & Benita, S. (2004). Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomedicine & Pharmacotherapy, 58(3), 173–182. doi:10.1016/j.biopha.2004.02.001
- Hu, J., Johnston, K. P., & Williams, R. O., III. (2004). Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs. Drug Development and Industrial Pharmacy, 30(3), 233–245. doi:10.1081/DDC-120030422
- Jain, N. K., & Gupta, U. (2008). Application of dendrimer–drug complexation in the enhancement of drug solubility and bioavailability. Expert Opinion on Drug Metabolism & Toxicology, 4(8), 1035–1052. doi:10.1517/17425255.4.8.1035
- Jones, M. C., & Leroux, J. C. (1999). Polymeric micelles – a new generation of colloidal drug carriers. European Journal of Pharmaceutics and Biopharmaceutics, 48(2), 101–111. doi:10.1016/S0939-6411(99)00039-9
- Kakkar, V., Singh, S., Singla, D., & Kaur, I. P. (2011). Exploring solid lipid nanoparticles to enhance the oral bioavailability of curcumin. Molecular Nutrition & Food Research, 55(3), 495–503. doi:10.1002/mnfr.v55.3
- Kanaujia, P., Ng, W. K., & Tan, R. B. (2014). Solid self-emulsifying drug delivery system (S-SEDDS) for improved dissolution rate of fenofibrate. Journal of Microencapsulation, 31(3), 293–298. doi:10.3109/02652048.2013.843601
- Kawabata, Y., Wada, K., Nakatani, M., Yamada, S., & Onoue, S. (2011). Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. International Journal of Pharmaceutics, 420(1), 1–10. doi:10.1016/j.ijpharm.2011.08.032
- Kawakami, K. (2012). Modification of physicochemical characteristics of active pharmaceutical ingredients and application of supersaturatable dosage forms for improving bioavailability of poorly absorbed drugs. Advanced Drug Delivery Reviews, 64(6), 480–495. doi:10.1016/j.addr.2011.10.009
- Ke, W., Zhao, Y., Huang, R., Jiang, C., & Pei, Y. (2008). Enhanced oral bioavailability of doxorubicin in a dendrimer drug delivery system. Journal of Pharmaceutical Sciences, 97(6), 2208–2216. doi:10.1002/jps.21155
- Khasia, H., & Khasia, V. (2012). International Journal of Pharmaceutical and Chemical Sciences, 1(1), 353.
- Kim, D. W., Kang, J. H., Oh, D. H., Yong, C. S., & Choi, H.-G. (2012). Development of novel flurbiprofen-loaded solid self-microemulsifying drug delivery system using gelatin as solid carrier. Journal of Microencapsulation, 29(4), 323–330. doi:10.3109/02652048.2011.651497
- Komal, S., Abdul, W., Habiba, A., Kinza, M., Maria, M., Waseem, K. M., … Din, F. U. (2017). Pharmaceutical Nanotechnology, 6, 2018.
- Krishnaiah, Y. S. (2012). Pharmaceutical Technologies for Enhancing Oral Bioavailability of Poorly Soluble Drugs. Journal of Bioequivalence & Bioavailability, 2.2, 028-036.
- Lee, S. C., Huh, K. M., Lee, J., Cho, Y. W., Galinsky, R. E., & Park, K. (2007). Hydrotropic polymeric micelles for enhanced paclitaxel solubility: In vitro and in vivo characterization. Biomacromolecules, 8(1), 202–208. doi:10.1021/bm060307b
- Li, H., Zhao, X., Ma, Y., Zhai, G., Li, L., & Lou, H. (2009). Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. Journal of Controlled Release, 133(3), 238–244. doi:10.1016/j.jconrel.2008.10.002
- Loftsson, T., Sigurđsson, H., Másson, M., & Schipper, N. (2004). Die Pharmazie – An International Journal of Pharmaceutical Sciences, 59(1), 25.
- Maria, M., Saba, I., Samreen, R., Ahmad, Z., Khan, G. M., Rehman, A., & Din, F. U. (2017). Nanoscale Research Letters, 12, 300. doi:10.1186/s11671-017-2046-4
- Millard, J. W., Alvarez-Nunez, F., & Yalkowsky, S. H. (2002). Solubilization by cosolvents. International Journal of Pharmaceutics, 245(1), 153–166. doi:10.1016/S0378-5173(02)00334-4
- Miller, D. A., McConville, J. T., Yang, W., Williams, R. O., & McGinity, J. W. (2007). Hot-melt extrusion for enhanced delivery of drug particles. Journal of Pharmaceutical Sciences, 96(2), 361–376. doi:10.1002/jps.20806
- Miroshnyk, I., Mirza, S., & Sandler, N. (2009). Pharmaceutical co-crystals – an opportunity for drug product enhancement. Expert Opinion on Drug Delivery, 6(4), 333–341. doi:10.1517/17425240902828304
- Mueller, E. A., Kovarik, J. M., van Bree, J. B., Tetzloff, W., Grevel, J., & Kutz, K. (1994). Pharmaceutical Research, 11(2), 301. doi:10.1023/A:1018923912135
- Muèller, R. H., Maèder, K., & Gohla, S. (2000). European Journal of Pharmaceutics and Biopharmaceutics, 50(1), 161. doi:10.1016/S0939-6411(00)00087-4
- Mura, P., Faucci, M. T., & Bettinetti, G. P. (2001). The influence of polyvinylpyrrolidone on naproxen complexation with hydroxypropyl-β-cyclodextrin. European Journal of Pharmaceutical Sciences, 13(2), 187–194. doi:10.1016/S0928-0987(01)00093-8
- Mutalik, S., Anju, P., Manoj, K., & Usha, A. N. (2008). Enhancement of dissolution rate and bioavailability of aceclofenac: A chitosan-based solvent change approach. International Journal of Pharmaceutics, 350(1), 279–290. doi:10.1016/j.ijpharm.2007.09.006
- Nidhi, K., Indrajeet, S., Khushboo, M., Gauri, K., & Sen, D. J. (2011). Hydrotropy: A promising tool for solubility Enhancement: A Review. International Journal of Drug Development and Research, 3(2), 26-33.
- Nigade, P. M., Patil, S. L., & Tiwari, S. S. (2012). International Journal of Pharmacy and Biological Sciences, 2(2), 42.
- Ober, C. A., & Gupta, R. B. (2012). Formation of itraconazole–succinic acid cocrystals by gas antisolvent cocrystallization. AAPS PharmSciTech, 13(4), 1396–1406. doi:10.1208/s12249-012-9866-4
- Otsuka, H., Nagasaki, Y., & Kataoka, K. (2003). PEGylated nanoparticles for biological and pharmaceutical applications. Advanced Drug Delivery Reviews, 55(3), 403–419. doi:10.1016/S0169-409X(02)00226-0
- Patel, J., Kevin, G., Patel, A., Raval, M., & Sheth, N. (2011). Design and development of a self-nanoemulsifying drug delivery system for telmisartan for oral drug delivery. International Journal of Pharmaceutical Investigation, 1(2), 112. doi:10.4103/2230-973X.82431
- Pitha, J., Milecki, J., Fales, H., Pannell, L., & Uekama, K. (1986). Hydroxypropyl-β-cyclodextrin: Preparation and characterization; effects on solubility of drugs. International Journal of Pharmaceutics, 29 (1), 73–82. doi:10.1016/0378-5173(86)90201-2
- Rapoport, N. (2007). Physical stimuli-responsive polymeric micelles for anti-cancer drug delivery. Progress in Polymer Science, 22(8–9), 962–990. doi:10.1016/j.progpolymsci.2007.05.009
- Rashid, R., Din, F. U., Mustapha, O., Kim, D. W., Yousaf, A. M., Park, J. H., … Choi, H. G. (2015). Effect of hydroxypropylcellulose and Tween 80 on physicochemical properties and bioavailability of ezetimibe-loaded solid dispersion. Carbohydrate Polymer, 130, 26–31. doi:10.1016/j.carbpol.2015.04.071
- Rashid, R., Kim, D. W., Yousaf, A. M., Mustapha, O., Din, F. U., Park, J. H., … Choi, H. G. (2015). International Journal of Nanomedicine, 10, 6147–6159.
- Rodriguez, L., Passerini, N., Cavallari, C., Cini, M., Sancin, P., & Fini, A. (1999). Description and preliminary evaluation of a new ultrasonic atomizer for spray-congealing processes. International Journal of Pharmaceutics, 183(2), 133. doi:10.1016/S0378-5173(99)00076-9
- Saenger, W. (1980). Cyclodextrin inclusion compounds in research and industry. Angewandte Chemie International Edition in English, 19(5), 344–362. doi:10.1002/(ISSN)1521-3773
- Salman, Q. O., Alam, Z., Muhammad, A., Sic, K. M., Ho, K. J., Seong, K. H., … Ki, K. J. (2016). European Journal O F Pharmaceutics and Biopharmaceutics, 108, 187–195. doi:10.1016/j.ejpb.2016.09.008
- Sauceau, M., Rodier, E., & Fages, J. (2008). Preparation of inclusion complex of piroxicam with cyclodextrin by using supercritical carbon dioxide. The Journal of Supercritical Fluids, 47(2), 326–332. doi:10.1016/j.supflu.2008.07.006
- Savjani, K. T., Gajjar, A. K., & Savjani, J. K. (2012). Drug solubility: Importance and enhancement techniques. ISRN Pharmaceutics, 2012. doi:10.5402/2012/195727
- Seedher, N., & Bhatia, S. (2003). Solubility enhancement of cox-2 inhibitors using various solvent systems. Aaps Pharmscitech, 4(3), 36–44. doi:10.1208/pt040333
- Sekhon, B. S. (2005). International Bulletin of Drug Research, 1(2), 24.
- Seo, A., & Schæfer, T. (2001). Melt agglomeration with polyethylene glycol beads at a low impeller speed in a high shear mixer. European Journal of Pharmaceutics and Biopharmaceutics, 52(3), 315–325. doi:10.1016/S0939-6411(01)00183-7
- Shah, N., Carvajal, M., Patel, C., Infeld, M., & Malick, A. (1994). Self-emulsifying drug delivery systems (SEDDS) with polyglycolyzed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. International Journal of Pharmaceutics, 106(1), 15–23. doi:10.1016/0378-5173(94)90271-2
- Shin, H. C., Alani, A. W., Rao, D. A., Rockich, N. C., & Kwon, G. S. (2009). Multi-drug loaded polymeric micelles for simultaneous delivery of poorly soluble anticancer drugs. Journal of Controlled Release, 140(3), 294–300. doi:10.1016/j.jconrel.2009.04.024
- Shiraki, K., Takata, N., Takano, R., Hayashi, Y., & Terada, K. (2008). Dissolution improvement and the mechanism of the improvement from cocrystallization of poorly water-soluble compounds. Pharmaceutical Research, 25(11), 2581–2592. doi:10.1007/s11095-008-9676-2
- Strickley, R. G. (2000). PDA-Journal of Pharmaceutical Science and Technology, 54(1), 69.
- Strickley, R. G. (2004). Solubilizing excipients in oral and injectable formulations. Pharmaceutical Research, 21(2), 201–230. doi:10.1023/B:PHAM.0000016235.32639.23
- Sun, S., & Zeng, H. (2002). Size-controlled synthesis of magnetite nanoparticles. Journal of the American Chemical Society, 124(28), 8204–8205. doi:10.1021/ja026501x
- Takahashi, M., Uechi, S., Takara, K., Asikin, Y., & Wada, K. (2009). Evaluation of an oral carrier system in rats: Bioavailability and antioxidant properties of liposome-encapsulated curcumin. Journal of Agricultural and Food Chemistry, 57(19), 9141–9146. doi:10.1021/jf9013923
- Talanov, V. S., Regino, C. A., Kobayashi, H., Bernardo, M., Choyke, P. L., & Brechbiel, M. W. (2006). Dendrimer-based nanoprobe for dual modality magnetic resonance and fluorescence imaging. Nano Letters, 6(7), 1459–1463. doi:10.1021/nl060765q
- Taneri, F., Ozcan, I., & Guneri, T. (2010). In vitro and in vivo evaluation of oral tablet formulations prepared with ketoconazole and hydroxypropyl- β-cyclodextrin. Drug Delivery, 17(3), 152–157. doi:10.3109/10717541003604890
- Tang, Y., Zhang, H., Lu, X., Jiang, L., Xi, X., Liu, J., & Zhu, J. (2015). Development and evaluation of a dry powder formulation of liposome-encapsulated oseltamivir phosphate for inhalation. Drug Delivery, 22(5), 608–618. doi:10.3109/10717544.2013.863526
- Thompson, D. O. (1997). Critical Reviews™ in Therapeutic Drug Carrier Systems, doi:10.1615/CritRevTherDrugCarrierSyst.v14.i1.10.
- Tomalia, D. A., & Frechet, J. M. (2002). Introduction to the dendritic state. Wiley Online Library, John Wiley & Sons Ltd Chichester UK. doi:10.1002/0470845821.ch1.
- Tomasik, P., & Schilling, C. H. (1998). Complexes of Starch with Inorganic Guests. Advances in Carbohydrate Chemistry and Biochemistry, 53, 263-345.
- ud Din, F., Aman, W., Ullah, I., Salman, Q. O., Mustapha, O., & Zeb, A. (2017). Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. International Joiurnal of Nanomedicine, 12, 7291–7309.
- ud Din, F. U., Cho, J. Y., Kim, D. W., Mustapha, O., Kim, D. S., RJ, T., … Choi, H. G. (2017). Drug Delivery, 24(1), 502–510. doi:10.1080/10717544.2016.1272651
- ud Din, F. U., Mustapha, O., & Kim, D. W. (2017). Acta Biomaterialia, 54, 239–248. doi:10.1016/j.actbio.2017.03.007
- ud Din, F. U., Mustapha, O., Kim, D. W., Rashid, R., Park, J. H., Choi, J. Y., … Choi, H.-G. (2015). Novel dual-reverse thermosensitive solid lipid nanoparticle-loaded hydrogel for rectal administration of flurbiprofen with improved bioavailability and reduced initial burst effect. European Journal of Pharmaceutics and Biopharmaceutics, 94, 64–72. doi:10.1016/j.ejpb.2015.04.019
- ud Din, F. U., Rashid, R., Mustapha, O., Kim, D. W., Park, J. H., Ku, S. K., … Yong, C. S. (2015). Development of a novel solid lipid nanoparticles loaded dual-reverse thermosensitive nanomicellefor intramuscular administration with sustained release and reduced toxicity. RSC Advances, 5, 43687.
- Üner, M., & Yener, G. (2007). International Journal of Nanomedicine, 2(3), 289.
- Vemavarapu, C., Mollan, M. J., Lodaya, M., & Needham, T. E. (2005). Design and process aspects of laboratory scale SCF particle formation systems. Journal of Pharmaceutics, 292(1), 1–16. doi:10.1016/j.ijpharm.2004.07.021
- Wakaskar, R. R. (2017a). Challenges Pertaining to Adverse Effects of Drugs. International Journal of Drug Development and Research, 9;01-02.
- Wakaskar, R. R. (2017b). Polymeric Micelles and their properties. Journal of Nanomedicine & Nanotechnology, 8(2), 1000433. doi:10.4172/2157-7439
- Wakaskar, R. R. (2017c). International Journal of Drug Development and Research, 9(3), 1–2.
- Wakaskar, R. R. (2018a). General overview of lipid–Polymer hybrid nanoparticles, dendrimers, micelles, liposomes, spongosomes and cubosomes. Journal of Drug Targeting, 26(4), 311–318. doi:10.1080/1061186X.2017.1367006
- Wakaskar, R. R. (2018b). Brief overview of nanoparticulate therapy in cancer. Journal of Drug Targeting, 26(2), 123–126. doi:10.1080/1061186X.2017.1347175
- Williams, H. D., Trevaskis, N. L., Charman, S. A., Shanker, R. M., Charman, W. N., Pouton, C. W., & Porter, C. J. (2013). Strategies to address low drug solubility in discovery and development. Pharmacological Reviews, 65(1), 315–499. doi:10.1124/pr.112.005660
- Xu, W., Ling, P., & Zhang, T. (2013). Polymeric Micelles, a promising drug delivery system to enhance bioavailability of poorly water-soluble drugs. Journal of Drug Delivery, 2013. doi:10.1155/2013/340315
- Yan, Y. D., Kim, J. A., Kwak, M. K., Yoo, B. K., Yong, C. S., & Choi, H. G. (2011). Enhanced oral bioavailability of curcumin via a solid lipid-based self-emulsifying drug delivery system using a spray-drying technique. Biological and Pharmaceutical Bulletin, 34(8), 1179–1186. doi:10.1248/bpb.34.1179
- Yang, T., Cui, F. D., Choi, M. K., Cho, J. W., Chung, S. J., Shim, C. K., & Kim, D. D. (2007). Enhanced solubility and stability of PEGylated liposomal paclitaxel: In vitro and in vivo evaluation. International Journal of Pharmaceutics, 338(1), 317–326. doi:10.1016/j.ijpharm.2007.02.011
- Zaheer, A., Naveen, M., Santosh, M. K., & Imran, K. (2011). Solubility enhancement of poorly water soluble drugs: A review. International Journal Of Pharmacy & Technology. 3 (1), 807–823.
- Zeb, A., Qureshi, O. S., Yu, C. H., Akram, M., Kim, H. S., Kim, M. S., … Kim, J. K. (2017). Enhanced anti-rheumatic activity of methotrexate-entrapped ultradeformable liposomal gel in adjuvant-induced arthritis rat model. International Journal of Pharmaceutics, 525(1), 92–100. doi:10.1016/j.ijpharm.2017.04.032
- Zhang, J. A., Anyarambhatla, G., Ma, L., Ugwu, S., Xuan, T., Sardone, T., & Ahmad, I. (2005). Development and characterization of a novel Cremophor EL free liposome-based paclitaxel (LEP-ETU) formulation. European journal o f pharmaceutics and biopharmaceutics. 59(1), 177-187.doi: 10.1016/j.ejpb.2004.06.009