Abstract
In the last ten years or so, attempts have been made to assess the structural properties of high solids foodstuffs using the WLF equation which has found a good degree of general acceptance in the investigation of amorphous synthetic polymers and diluted systems. However, there appears to be a misguided effort to apply the equation in a variety of molecular processes ‘as long as it fits’. It should be remembered that the merit of the WLF scheme is invariably associated with the theory of free volume. The theory follows the kinetic contributions to polymer relaxation at the glass transition region, which is a second order thermodynamic process thus signifying a change in state not in phase. Critical application of the combined WLF/free volume theoretical framework to high sugar/biopolymer mixtures using the technique of small-deformation dynamic oscillation yields the rheological glass transition temperature (T g ), the thermal expansion coefficient (α f ) and the fractional free volume (f g ) at T g . The physical significance of the rheological T g lies in providing a threshold above which the free volume effects are superseded by the predictions of the reaction-rate theory. Furthermore, the treatment is capable of resolving the complicated mechanical properties of high solids foodstuffs into one basic function of frequency (time) alone and one basic function of temperature alone. The last section of the article deals with the concept of the monomeric friction coefficient used in the physics of synthetic materials to relate measurable viscoelastic constants to molecular characteristics. It remains to be seen whether a similar advance can be achieved in biological melts and glasses.
OBJECTIVES OF THE REVIEW
The concepts of this manuscript are presented in four sections thus aiming to i) emphasize the utility of the Williams, Landel and Ferry equation in association with the theory of free volume in vitrification phenomena, as opposed to its use as a mere exponential fit; ii) document the applicability of this equation to the viscoelasticity of biological glasses thus offering a definition for the rheological glass transition temperature based on the free volume theory; iii) offer a theoretical foundation of the superposition of mechanical data by separating them into a basic function of time alone and a basic function of temperature alone and iv) correlate the structural properties of materials to molecular weight and conformation characteristics using the concept of monomeric friction coefficient.
What happens in very high sugar/biopolymer environments has largely been ignored by the research community, yet this is of great importance for the confectionery industry and has other applications, for example, flavour encapsulation and preservation of bioactive molecules in glassy carbohydrate matrices. It is our view, that such a fundamental approach can tackle this under researched area thus inspiring the inception of a number of potential applications in the food industry.
THE SUBTLETY OF USING THE WLF/FREE VOLUME THEORY IN THE VITRIFICATION OF HIGH SOLIDS SYSTEMS
The Concept of Free Volume
Back in 1957, Doolittle and Doolittle were interested in the Newtonian viscosity of the liquid n-alkanes (C5 to C64) over a wide range of temperatures. According to custom, the simple Andrade temperature equation (1930):
Advances in microcomputing in the last twenty years or so has allowed the rapid development of the technique of dynamic oscillation, with computer driven rheometers becoming commonplace in R&D laboratories. These are now established as a productive line of attack for the understanding of function-structure-texture relationships in food products. The technique is capable of resolving the structural properties of foodstuffs into a solid and a liquid-like response (G′ and G″, respectively). In the former, the applied stress is proportional to the generated strain with the oscillatory waves of stress and strain being in phase with each other. For a viscous system, however, the stress will be proportional to the strain-rate at any given time and thus the maximum stress will occur when the slope of the strain wave is a maximum, i.e. at the zero cross-over points. This results in a phase-shift of π/2 radians. A foodstuff will therefore produce an overall response (G *) having contributions from both the in-phase and out-of-phase components in the following form (Richardson and Kasapis, 1998):
The WLF Equation
In the absence of a phase transition, viscoelastic moduli at two different temperatures can be related with the following expression:
The glass transition temperature is now defined as the point where the thermal expansion coefficient, α f , undergoes a discontinuity and below which thermal motions become extremely slow (Ferry, 1980). Substituting equation Equation6 in equation Equation5 and implementing a straightforward calculation gives:
The approach has proved to be widely applicable and three decades after its introduction, Sperling (1986) acknowledged that ‘for a generation of synthetic polymer scientists and rheologists the WLF equation has proved a mainstay both in utility and theory’. In the last ten years or so, there has been an appreciation that much of the work in amorphous synthetic polymers and diluted systems is applicable to biological rubbers and glasses (Slade and Levine, 1991). However, the paper by Lopes da Silva and co-workers (1994) demonstrated that the WLF scheme is not applicable to the aqueous dispersions of the pectin polysaccharide due to the microaggregate-induced heterogeneity in the gel. Thus the authors were able to show that ‘a smooth master curve could not be obtained for both moduli simultaneously or for each one individually ⃜satisfactory reduction of the data to a single curve was not obtained, irrespective of the frequency shift factor used, for each modulus individually or with vertical shift factors higher than those calculated by the experimental temperature-density factor’.
DMTA Studies on Starches
Vitrification studies were also carried out in high-solid polysaccharide systems using mainly the technique of dynamic mechanical thermal analysis (DMTA). Figure illustrates the effect of water content and type of sugar on the structural properties of waxy maize starch. There is an order of magnitude drop in the heating profiles of storage (E′) and loss (E″) Young's modulus in the presence of 10.5% water which has been attributed to the glass transition of amylopectin (Kalichevsky et al., 1992). The plasticizing effect of water was confirmed by increasing its content to ∼19% thus moving the vitrification spectrum of the amylopectin/fructose mixture to lower temperatures (Kalichevsky and Blanshard, 1993). However, the order of magnitude change in material properties indicating glass transition is unusually low, i.e. between one and two orders of magnitude for the amylopectin and the amylopectin/fructose samples, respectively.
Figure 1. DMTA heating of amylopectin with 10.5% water from 20 to 130°C (circles), and a second heating run from −80 to 80°C for amylopectin with 25% fructose and ∼19% water shown in squares (Kalichevsky et al., 1992; Kalichevsky and Blanshard, 1993).
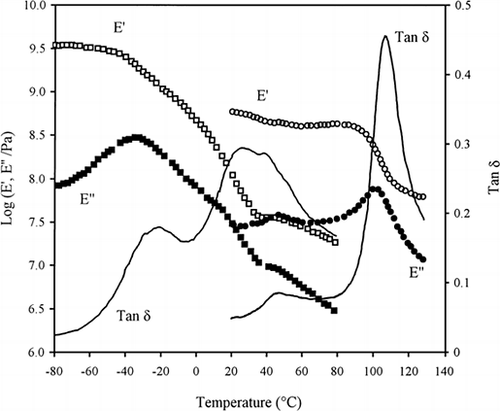
Favier et al. (1995) prepared suspensions of ordered cellulose whiskers in a lattice of co-polymerised styrene and butyl acrylate, and Thompson and Woods (1956) cooled down slowly polyethylene terephthalate. The outcome of tensile measurements on these materials was a reduction in the magnitude of the glass transition from four to one decades of modulus with increasing order in the system. Sample bars for DMTA analysis are made by pressing the powders at about 108 Pa in the presence of limited levels of water thus preventing the polysaccharide from reaching a state of molecular hydration. It appears that the lack of hydration of the carbohydrate chains creates structures with excessive molecular order, which diminishes the development of glass transitions in the system. Consequently, the values of tan δ (E″/E′) in Figure remain well below unity which is a sign of limited vitrification phenomena as a function of temperature.
In the case of the amylopectin/fructose mixture, the appearance of two tan δ peaks indicates that some of the co-solute forms a separate phase which is not mixed on a molecular level with amylopectin thus not acting as the polymer's plasticizer. This makes inappropriate the application of the Couchman-Karasz equation (1978) which requires molecularly compatible polymer-plasticiser-solvent mixtures. The preponderance of low-mobility ordered regions in the samples prepared for DMTA analysis is also reflected in a constant energy of activation obtained using the Arrhenius plot for first order thermodynamic transitions (Kalichevsky et al., 1993). This implies a change in phase and argues for the absence of free volume effects modeled by the WLF equation (discussed above).
The glass transition is considered to be a second order thermodynamic transition in which the material undergoes a change in state but not in phase. Experimentally, this is supported by calorimetric studies on supercooled glycerol which produced a step-change in heat capacity as a function of temperature at 190 K (Allen, 1993). The polydisperse nature of food materials results in structural changes occurring over a wide temperature range. The resulting curve is an array of minute step changes reflecting the different glass transition temperatures (T g ) of the molecular fractions of the sample. In calorimetric studies, empirical indices of T g have been adopted at various positions of the thermal event. Similarly, the glass transition temperature (T g ) in Figure has been taken as the maximum in the values of tan δ but in the absence of a distinct molecular process associated with this point, the approach also denotes an empirical index.
USING THE METHOD OF REDUCED VARIABLES IN THE PREDICTION OF THE GLASSS TRANSITION TEMPERATURE
Treating Dynamic Oscillatory Data with the Method of Reduced Variables
Research on amorphous synthetic polymers, e.g. poly(n-octyl methacrylate), demonstrated that in the transition zone between the rubber and glass-like consistency the temperature dependence of viscoelastic properties is quite spectacular (Dannhauser et al., 1958). At the glass transition temperature, the only residual contraction is of solid-like character which reduces dramatically the diffusion rate of water molecules and compounds needed to support chemical reactions (Slade and Levine, 1993; Mitchell, 1998). Thus the physical state of foodstuffs becomes very stable and the concept of T g allows prediction of physicochemical properties over an extended time scale of storage.
Viscosity is the simplest rheological parameter to measure and its temperature dependence has been used in the past to estimate the T g of materials. Angell (1988) reproduced viscosity values for inorganic melts and organic liquids achieving 1012 Pa s at the glassy state. The approach is valid provided that Newtonian liquids are analysed or the shear thinning properties of the material are well mapped out. However, the paper on the vitrification behaviour of maltose-water mixtures demonstrates the difficulty of experimenting at conditions of extreme sample rigidity (Noel et al., 1991). Viscosities values did not exceed 106.5 Pa s and a long extrapolation to 1012 Pa s was implemented in an attempt to predict the value of T g . Similarly, the temperature dependence of viscosity for honey was recorded with a view to predicting its glass transition temperature from relatively low viscosity values (below 2.4 kPa s in Bhandari et al., 1999). It was also stated that the ‘T g can be a useful tool to predict crystallisation behavior of honey’ but, in our view, these are fundamentally different molecular processes and should not be referred to interchangeably.
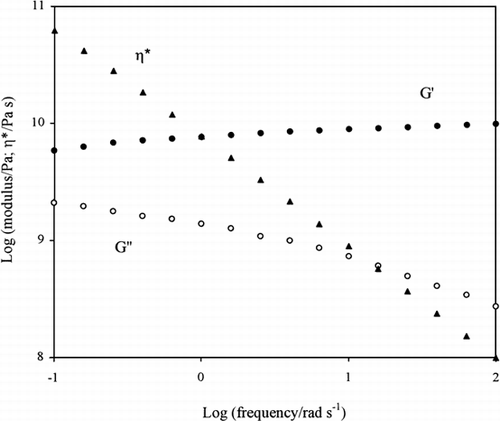
The textural properties of high sugar preparations in the presence of protein or polysaccharide at levels of normal industrial use (below 5% and 1%, respectively) are recently enjoying increasing attention from researchers and product developers alike, and there is a need to develop useful theoretical and empirical relationships. Figure illustrates a typical mechanical spectrum of the glassy state covering a frequency range from 0.1 to 100 rad/s (Kasapis et al., 1999). The biological glass is a mixture of 5% acid pigskin gelatin (Mn = 68,000), 30% sucrose and 50% glucose syrup (dextrose equivalent 42). Within the accessible range of observation, the values of G′ lie above those of G″ (solid-like behaviour) and both moduli are relatively independent of the frequency of oscillation (ω). However, the log of the complex viscosity (η*) descends steeply from almost 1011 to 108 Pa s as a function of log ω. Thus the absence of a ‘plateau’ in the trace of η* makes predictions of T g from viscosity readings rather tenuous.
Figure 2. Frequency sweep of storage modulus, loss modulus, and complex viscosity for 5% acid pigskin gelatin in the presence of 30% sucrose and 50% glucose syrup obtained at −55°C and a strain of 0.001% (Kasapis et al., 1999).
Issues Pertaining to the Rheological Glass Transition Temperature
Following the synthetic polymer approach (Child and Ferry, 1957), we attempted to pinpoint the T g of higher sugar/biopolymer mixtures by means of measurements of shear moduli within the linear viscoelastic region. Figure depicts the composite curve (also known as master curve) obtained for a mixture of 25% acid pigskin gelatin (Mn = 29,200), 40% sucrose and 15% glucose syrup (dextrose equivalent 42). The curve covers almost fifteen decades of frequency and six orders of magnitude of shear modulus. Normal experimental procedure covers three or four decades of frequency (Whorlow, 1992). To achieve a wider frequency window, we heated the preparation from −55 to 14°C at a scan rate of 1°C/min and obtained frequency sweeps between 0.1 and 100 rad/s at constant temperature intervals of three degrees centigrade. A reference temperature was chosen arbitrarily (−25°C) within the glass transition region and the remaining mechanical spectra were shifted horizontally along the log frequency axis until they fell into a single curve.
Figure 3. Composite curve of storage and loss modulus for a mixture of 25% acid pigskin gelatin with 40% sucrose and 15% glucose syrup. The reference temperature is −25°C (Kasapis et al., submitted).
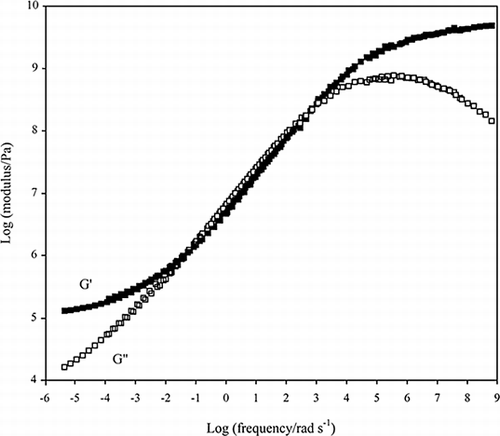
The scheme for construction of a composite curve by empirical shifts of data is known as the method of reduced variables or the time-temperature superposition principle (TTS in Tobolsky, 1956). Commonly, mechanical spectra are obtained over a wide temperature range of 30 to 70 degrees centigrade, which reflects the glass transition region. Its applicability requires exact matching of the shapes of experimental curves. The extent of shifting is reflected in the values of the shift factor (a T in equation Equation4) which should be the same for both the G′ and G″ traces. At the bottom range of frequency, part of the rubbery region is unveiled with the storage modulus being dominant (Figure ). This is followed by the glass transition region where configurational rearrangements of regions of the molecule contribute mainly to an energy dissipating process and the sample exhibits an overall liquid-like response (G″ > G′). At the upper range of frequency, G′ overtakes G′ once more and the sample enters the glassy state where only stretching and bending of chemical bonds are accomplished (Ferry, 1980a).
The set of shift factors obtained for the temperature range of the glass transition region in Figure was plotted using the linearised form of equation Equation7, thus calculating the C 1 0 and C 2 0 from the gradient and the intercept of the linear fit, respectively. The rheological T g for the mixture of 25% gelatin with 55% co-solute was found to be −39°C (Table ) taking into account that T g − 50 = T 0 − C 2 0 (Ferry, 1980). The range of temperatures T g + 53°C demarcates the glass transition region from the end of the rubbery plateau (≈ 14°C) to the onset of the glassy state (−39°C) where the kinetics of the free volume theory are fully operative. The WLF equation holds for any temperature within the glass transition region including T g (Ward and Hadley, 1993), a result which leads to the following relation:
Table 1. Parameters Characterising the Temperature Dependence of a T for the Sugar/Biopolymer Systems
In conclusion, this work argues that addition of small amounts of biopolymer to a sugar preparation alters dramatically the glass transition temperature. The acceleration of vitrification properties of sugar in the presence of κ-carrageenan or gellan should be due to the development of strong interactions between the polysaccharide and the surrounding high-solids environment leading to molecular immobilization and early vitrification. This is assisted by the presence of 0.01 M KCl and 0.007 N Ca2+ (κ-carrageenan and gellan, respectively), which stabilise the cross-links of the polysaccharide network (Table ). In the absence of added counterions, the vitrification process of κ-carrageenan/sugar and gellan/sugar follows a slow temperature dependence seen for single sugar preparations. In the absence of biopolymer, the values of the rheological T g are congruent with those reported for the calorimetric T g (Sworn and Kasapis, 1998; Roos, 1993). In the presence of biopolymer, however, calorimetry produces T g values which are dominated by the sugar spectrum (Slade and Levine, 1991; Goff et al., 1993; Blond, 1994). This discrepancy invites a debate as to the true molecular nature of the glass transition temperature measured by the two techniques. Finally, the estimates of T g obtained by treating a set of frequency sweeps taken at different temperatures with the WLF equation, are reproduced with an accuracy of ±2 degrees centigrade by the cooling/heating profiles of the same samples as the cross over of G′ and G″ traces passing from the glass transition region to the glassy state and, hence, are experimentally verified (Evageliou et al., 1998; Kasapis et al., 1999). The massive changes in the nature or the glassy behaviour of sugar cannot be explained on the basis of the simple mixing rule implemented by the Couchman-Karasz equation (1978) but should be the result of molecular interactions between the polymer and co-solute at high levels of solids (>70%).
In the first application of the WLF equation, average values of C1 g (17.44) and C2 g (51.6) were used to fit the data, with the authors acknowledging the approximate nature of this calculation owing to the uncertainty in specifying T g and the difficulty of experimenting near T g (Williams et al, 1955). In a somewhat better approximation, values of C1 0 (8.86) and C2 0 (101.6) were used but, twenty five years later, Ferry (1980) pointed out that ‘the actual variation from one polymer to another is too great to permit use of these universal values’. Recent examination of the WLF equation with universal and variable coefficients reconfirmed the inability of the former to describe the pattern of vitrification phenomena at temperature ranges starting about 20 to 30°C above the glass transition temperature (Peleg, 1992). This is also evident from the corresponding values in Table for the vitrification properties of high sugar/biopolymer mixtures. In the absence of other specific data, the universal values were considered for the determination of the time dependence of lactose and sucrose crystallisation (Roos and Karel, 1991). The experimental temperature range was rather limited (20 degrees) and allowed simultaneous application of both the Arrhenius} and WLF fits, but the application of the latter is questionable since its physical significance is confined to free-volume phenomena (Nelson and Labuza, 1994).
SEPARATION OF THE VARIABLES OF TEMPERATURE AND FREQUENCY
The Basic Function of Temperature
Determination of T g constitutes the first step in fingerprinting the process of vitrification of a material. Qualitatively, this is accompanied by an experimental procedure which examines the variation of shear or Young's modulus as a function of temperature at a constant frequency (see, for example, the thermal profiles in Figure ). A more valuable find, however, would allow complete dissociation of the contributions of frequency and temperature to the overall mechanical behaviour in the form of a basic function of time alone and a basic function of temperature alone (Ferry and Fitzgerald, 1954).
In synthetic polymer research, the former, in the region of short times, is roughly similar for all polymers. In the region of long times, it depends primarily on molecular weight distribution. The latter depends primarily on chemical characteristics of the polymer and, if diluent is present, its nature and concentration. The separation is based on the assumption that during a change in state all relaxation times of a molecular process depend identically on temperature. In practice, the effect of changing the temperature on G * is to shift the frequency scale of mechanical spectra, taken in a sequence of temperature intervals, in the manner discussed in Figure . This makes the shift factor in equation Equation4 a fundamental descriptor of the temperature dependence of viscoelastic functions.
In Figure a, the factor a T is plotted against the temperature range of the glass transition region for the composition of 25% gelatin + 40% sucrose + 15% glucose syrup. The WLF equation represents a good fit of the empirically derived values of a T as a function of temperature in the glass transition region thus determining how much the frequency scale shifts with temperature. As discussed in the preceding section, the approach predicts the value of T g which is −39°C. The WLF equation is unable to follow progress in mechanical properties at the lower temperatures of the glassy state (down to −55°C), which are better described by the mathematical expression of Andrade (Figure b). This yields the concept of activation energy (E a ) for an elementary flow process in the glassy state which is independent of temperature. Similar transformation from free-volume derived effects in the glass transition region to the process of an energetic barrier to rotation in the solid-like environment of the glassy state was observed for high sugar/κ-carrageenan, gellan, locust bean gum and guar gum mixtures. The phenomenon appears to be universal, thus prompting us to assign physical significance to the rheological T g as the threshold of two distinct molecular processes (Evageliou et al., 1998; Sworn and Kasapis, 1998; Kasapis et al., 1999; Kasapis et al., 2000; Kasapis and Al-Marhoobi, 2000).
Figure 4. The effect of temperature on the extent of empirical shifting, a T , which resulted in the construction of the composite curve in Figure . The temperature function was characterised (a) by the WLF fit in the glass transition and (b) the linear Arrhenius fit in the glassy state (Kasapis et al., submitted).
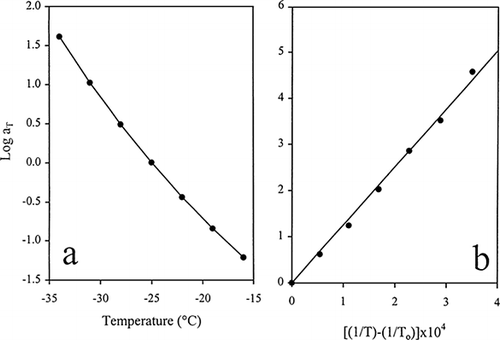
The temperature function in the glass transition can be compared directly with the activation energy in the glassy state using a modification of the WLF equation. This gives rise to the energy of vitrification (E v ) which is associated with the difficulty for transverse string-like vibrations over several molecules to occur. There is a certain advantage in using E v since it is independent of an arbitrary choice of reference temperature which determines the magnitude of factor a T . Thus by differentiating the WLF equation we obtain (Ferry and Fitzgerald, 1954):
First Approximation Calculations in the Time Function
Regarding the time function, the composite spectrum in Figure provides a basic understanding of the pattern of mechanical relaxation in amorphous systems (frequency (Hz) = 1/time (s)). For comparisons between several polymers, however, the distribution function of relaxation times, Φ, is preferred because it can be derived from both dynamic and transient measurements (Fitzgerald and Ferry, 1953; Bischoff et al., 1952). Furthermore, the function connects together the values of G′ and G″ although they are independent at a single frequency of measurement (Ferry and Williams, 1952):
The upper graphs in Figure illustrate the outcome of such calculations carried out using small deformation dynamic data on 0.7% agarose + 50% sucrose + 35% glucose syrup (A) and 0.82% deacylated gellan + 40% sucrose + 40% glucose syrup (G). In the former, traces level off at long timescales of measurement thus signifying formation of a permanent network within the experimental constraints (Tsoga et al., 1999). In the absence of added counterions, gellan chains are unable to hold the system together with the curves showing a steep portion characteristic of flow at times longer than 103 s (Sworn and Kasapis, 1999). In both cases, reduction in the timescale of observation produces a dramatic increase in the values of shear modulus, a result that signifies the advent of vitrification phenomena.
Figure 5. Logarithmic effect of relaxation times (τ) on the relaxation function (Φ) obtained from reduced mechanical spectra of G′ and G′ for high sugar agarose (A) and deacylated gellan (G). Upper and lower graphs are first-approximation and second-approximation calculations, respectively. Dashed lines give the slope predicted by the Rouse theory (Tsoga et al., 1999; Sworn and Kasapis, 1999; Kasapis and Sworn, 2000; Kasapis and Sablani, 2000).
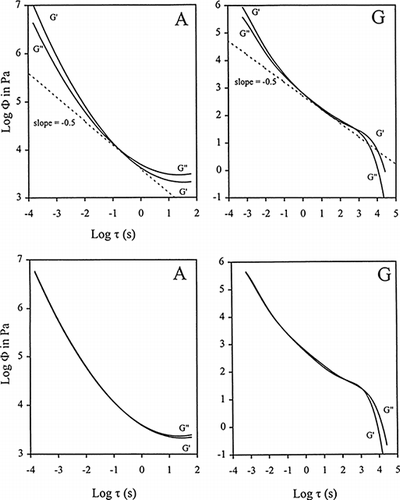
Second Approximation Calculations in the Time Function
Storage and loss modulus data is used to estimate the distribution function but the treatment fails to harmonize the derived curves at both ends of the experimental window of observation. However, values converge at a line with a theoretical slope of −0.5 where the relaxation spectrum is the outcome of fairly long-range cooperative motions of the chain backbone (Rouse, 1953). The need to minimize the discrepancy between the two predictions of the shear moduli led to the development of second approximation methods in the derivation of the mechanical relaxation distribution. The refinement was based on the observation that the Φ curves rise at short times with slopes being a simple power function of τ (in Figure and Ferry et al., 1951). Thus the analytical form Φ = kτ−m was assumed and then substituted in the ‘Maxwellian equations’ of shear modulus to give (Ferry et al., 1952):
THE MONOMERIC FRICTION COEFFICIENT IN THE GLASS TRANSITION ZONE
The Concept of Monomeric Friction Coefficient
Estimation of the glass transition temperature and the time/temperature function paves the way for correlating the measurable viscoelastic parameters with molecular characteristics. This has been a major goal in the physics of synthetic melts and glasses, and is based on the observation that viscoelastic quantities are governed by the dimensions of a macromolecule and the density dependent friction factor (Fox and Loshaek, 1955; Kraus and Gruver, 1965). For meaningful comparisons between different systems, the frictional resistance per monomer unit encountered by a chain segment is considered which constitutes the monomeric friction coefficient, ζ o . The resistance to segment relaxation is enhanced by local and long range forces in the form of intrachain and interchain physical bonds and topological entanglements. This increases the magnitude of ζ o , which is calculated by the relaxation spectrum Φ as follows (Ferry and Landel, 1956):
The Effect of Molecular Weight on Fractional Free Volume and Monomeric Friction Coefficient
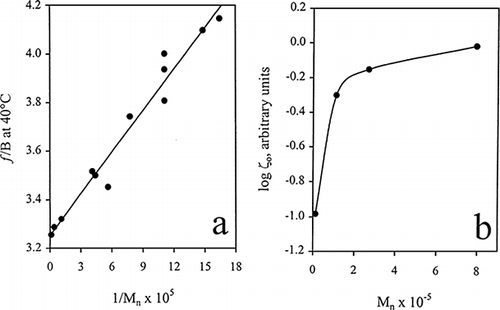
Work on polyvinyl acetate fractions and blends demonstrates that ζ o increases with molecular weight and eventually reaches a constant limiting value ζ oo (Ziabicki and Klonowski, 1975). To rationalise this behaviour, it is considered that the fractional free volume is a linear function of molecular weight in the following form (Fox and Flory, 1950):
Figure a depicts the application of equation Equation15 to polyvinyl acetate data yielding a value for the constant A of 52 g/mole at 40°C (Ninomiya et al., 1963). This value is related to the dimension of the polymer chain with low molecular weights imparting an additional free volume due to imperfect packing around the ends of the molecule. In the case of polydisperse biological materials, the approach should be used in conjunction with the number average molecular weight (M n ) which is inversely proportional to the number of chains ends. At infinite M n , a value for f oo is obtained from the intercept of the linear fit in Figure a. For the sake of simplicity, B has usually been set arbitrarily equal to unity. This implies that a f is more nearly equal to Δa, i.e. the difference between the magnitudes of a l and a g , which are characteristic of the thermal expansion coefficients of liquids and glasses, respectively (Ferry, 1980).
Figure 6. The effect of number average molecular weight of polyvinyl acetate on (a) fractional free volume and (b) the monomeric friction coefficient at 40°C (Ninomiya et al., 1963; Ziabicki and Klonowski, 1975).
As for the viscoelastic functions, equation Equation5 can be used to derive an analogue for the monomeric friction coefficient (Ninomiya and Ferry, 1963):
Acknowledgments
REFERENCES
- Allen , G. A. 1993 . “ History of the Glassy State ” . In The Glassy State in Foods Edited by: Blanshard , J. M.V. and Lillford , P. J. 1 – 12 . Nottingham : Nottingham University Press .
- Al-Ruqaie , I. , Kasapis , S. , Richardson , R. K. and Mitchell , G. 1997 . The Glass Transition Zone in High Solids Pectin and Gellan Preparations . Polymer , 38 : 5685 – 5694 .
- Andrade , E. N. and da , C. 1930 . The Viscosity of Liquids . Nature , 125 : 309 – 310 .
- Angell , C. A. 1988 . Perspective on the Glass Transition . J. Phys. Chem. Solids , 49 : 863 – 871 .
- Berry , G. C. and Fox , T. G. 1968 . The Viscosity of Polymers and their Concentrated Solutions . Advances in Polymer Science , 5 : 261 – 357 .
- Bhandari , B. , D'Arcy , B. and Kelly , C. 1999 . Rheology and Crystallization Kinetics of Honey: Present Status . International Journal of Food Properties , 2 : 217 – 226 .
- Bischoff , J. , Catsiff , E. and Tobolsky , A. V. 1952 . Elastoviscous Properties of Amorphous Polymers in the Transition Region. I . Journal of the American Chemical Society , 74 : 3378 – 3381 .
- Blond , G. 1994 . Mechanical Properties of Frozen Model Solutions . Journal of Food Engineering , 22 : 253 – 269 .
- Child , W. C. Jr. and Ferry , J. D. 1994 . Dynamic Mechanical Properties of Poly-n-butyl Methacrylate . Journal of Colloid Science , 12 : 327 – 341 .
- Couchman , P. R. and Karasz , F. E. 1978 . A Classical Thermodynamic Discussion of the Effect of Composition on Glass Transition Temperatures . Macromolecules , 11 : 117 – 119 .
- Dannhauser , W. , Child , W. C. Jr. and Ferry , J. D. 1958 . Dynamic Mechanical Properties of Poly-n-octyl Methacrylate . Journal of Colloid Science , 13 : 103 – 113 .
- Doolittle , A. K. and Doolittle , D. B. 1957 . Studies in Newtonian Flow. V. Further Verification of the Free-Space Viscosity Equation . Journal of Applied Physics , 28 : 901 – 905 .
- Evageliou , V. , Kasapis , S. and Hember , M. W.N. 1998 . itrification of κ-Carrageenan in the Presence of High Levels of Glucose Syrup . Polymer , 39 : 3909 – 3917 .
- Favier , V. , Chanzy , H. and Cavaillé , J. Y. 1995 . Polymer Nanocomposites Reinforced by Cellulose Whiskers . Macromolecules , 28 : 6365 – 6367 .
- Ferry , J. D. 1980 . “ Dependence of Viscoelastic Behavior on Temperature and Pressure ” . In Viscoelastic Properties of Polymers 264 – 320 . New York : John Wiley .
- Ferry , J. D. 1980a . “ Molecular Theory for Undiluted Amorphous Polymers and Concentrated Solutions; Networks and Entanglements ” . In Viscoelastic Properties of Polymers 224 – 263 . New York : John Wiley .
- Ferry , J. D. and Fitzgerald , E. R. 1954 . “ Dynamic Rheological Properties of Linear Polymers ” . In Proceedings of the Second International Congress on Rheology Edited by: Harrison , V. G.W. 140 – 149 . London : Butterworths Scientific .
- Ferry , J. D. and Landel , R. F. 1956 . Molecular Friction Coefficients in Polymers and their Temperature Dependence . Kolloid-Zeitschrift , 148 : 1 – 6 .
- Ferry , J. D. and Williams , M. L. 1952 . Second Approximation Methods for Determining the Relaxation Time Spectrum of a Viscoelastic Material . Journal of Colloid Science , 7 : 347 – 353 .
- Ferry , J. D. , Landel , R. F. and Williams , M. L. 1955 . Extensions of the Rouse Theory of Viscoelastic Properties to Undiluted Linear Polymers . Journal of Applied Physics , 26 : 359 – 362 .
- Ferry , J. D. , Fitzgerald , E. R. , Grandine , L. D. Jr. and Williams , M. L. 1952 . Temperature Dependence of Dynamic Properties of Elastomers; Relaxation Distributions . Industrial and Engineering Chemistry , 44 : 703 – 706 .
- Ferry , J. D. , Fitzgerald , E. R. , Johnson , M. F. and Grandine , L. D. Jr. 1951 . Mechanical Properties of Substances of High Molecular Weight. X. The Relaxation Distribution Function in Polyisobutylene and its Solutions . Journal of Applied Physics , 22 : 717 – 722 .
- Ferry , J. D. , Sawyer , W. M. , Browning , G. V. and Groth , A. H. Jr. 1950 . Mechanical Properties of Substances of High Molecular Weight. VIII. Dispersion of Dynamic Rigidity and Viscosity in Concentrated Polyvinyl Acetate Solutions . Journal of Applied Physics , 21 : 513 – 517 .
- Fitzgerald , E. R. and Ferry , J. D. 1953 . Method for Determining the Dynamic Mechanical Behavior of Gels and Solids at Audio-Frequencies; Comparison of Mechanical and Electrical Properties . Journal of Colloid Science , 8 : 1 – 34 .
- Fox , T. G. Jr. and Flory , P. J. 1950 . Second-Order Transition Temperatures and Related Properties of Polystyrene. I. Influence of Molecular Weight . Journal of Applied Physics , 21 : 581 – 591 .
- Fox , T. G. Jr. and Loshaek , S. 1955 . Isothermal Viscosity-Molecular Weight Dependence for Long Polymer Chains . Journal of Applied Physics , 26 : 1080 – 1082 .
- Goff , H. D. , Caldwell , K. B. and Stanley , D. W. 1993 . The Influence of Polysaccharides on the Glass Transition in Frozen Sucrose Solutions and Ice Cream . Journal of Dairy Science , 76 : 1268 – 1277 .
- Kalichevsky , M. T. and Blanshard , J. M.V. 1993 . The Effect of Fructose and Water on the Glass Transition of Amylopectin . Carbohydrate Polymers , 20 : 107 – 113 .
- Kalichevsky , M. T. , Blanshard , J. M.V. and Marsh , R. D.L. 1993 . “ Applications of Mechanical Spectroscopy to the Study of Glassy Biopolymers and Related Systems ” . In The Glassy State in Foods Edited by: Blanshard , J. M.V. and Lillford , P. J. 133 – 156 . Nottingham : Nottingham University Press .
- Kalichevsky , M. T. , Jaroszkiewicz , E. M. , Ablett , S. , Blanshard , J. M.V. and Lillford , P. J. 1992 . The Glass Transition of Amylopectin Measured by DSC, DMTA and NMR . Carbohydrate Polymers , 18 : 77 – 88 .
- Kasapis , S. and Al-Marhoobi , I. M.A. 2000 . “ Glass Transitions in High Sugar/Biopolymer Mixtures–Some Recent Developments ” . In Gums and Stabilisers for the Food Industry 10 Edited by: Williams , P. A. and Phillips , G. O. 303 – 313 . Cambridge : The Royal Society of Chemistry .
- Kasapis , S. and Sablani , S. S. 2000 . First and Second Approximation Calculations in the Relaxation Function of High Sugar/Polysaccharide Systems . International Journal of Biological Macromolecules , 27 : 301 – 305 .
- Kasapis , S. and Sworn , G. 2000 . Separation of the Variables of Time and Temperature in the Mechanical Properties of High Sugar/Polysaccharide Mixtures . Biopolymers , 53 : 40 – 45 .
- Kasapis , S. , Al-Marhoobi , I. M.A. and Giannouli , P. 1999 . Molecular Order Versus Vitrification in High Sugar Blends of Gelatin and κ-Carrageenan . Journal of Agricultural and Food Chemistry , 47 : 4944 – 4949 .
- Kasapis , S. , Al-Marhoobi , I. M.A. and Khan , A. J. 2000 . Viscous Solutions, Networks and the Glass Transition in High Sugar Galactomannan and κ-Carrageenan Mixtures . International Journal of Biological Macromolecules , 27 : 13 – 20 .
- Kasapis , S. , Al-Marhoobi , I. M.A. and Mitchell , J. R. 2000 . Molecular weight effects on the glass transition of gelatin/co-solute mixtures Macromolecules submitted
- Kraus , G. and Gruver , J. T. 1965 . Rheological Properties of Multichain Polybutadienes . Journal of Polymer Science , 3A : 105 – 122 .
- Landel , R. F. and Ferry , J. D. 1956 . Dynamic Mechanical Properties of the System Cellulose Tributyrate-Dimethyl Phthalate . Journal of Physical Chemistry , 60 : 294 – 301 .
- Lopes da Silva , J. A. , Gonçcalves , M. P. and Rao , M. A. 1994 . Influence of Temperature on the Dynamic and Steady-Shear Rheology of Pectin Dispersions . Carbohydate Polymers , 23 : 77 – 87 .
- Mitchell , J. R. 1998 . “ Water and Food Macromolecules ” . In Functional Properties of Food Macromolecules Edited by: Hill , S. E. , Ledward , D. A. and Mitchell , J. R. 50 – 76 . Gaithersburg : Aspen Publishers .
- Nelson , K. A. and Labuza , T. P. 1994 . Water Activity and Food Polymer Science: Implications of State on Arrhenius and WLF Models in Predicting Shelf Life . Journal of Food Engineering , 22 : 271 – 289 .
- Ninomiya , K. and Ferry , J. D. 1963 . Viscoelastic Properties of Polyvinyl Acetates. I. Creep Studies of Fractions . Journal of Physical Chemistry , 67 : 2292 – 2296 .
- Ninomiya , K. , Ferry , J. D. and Õyanagi , Y. 1963 . Viscoelastic Properties of Polyvinyl Acetates. II. Creep Studies of Blends . Journal of Physical Chemistry , 67 : 2297 – 2308 .
- Noel , T. R. , Ring , S. G. and Whittam , M. A. 1991 . Kinetic Aspects of the Glass-Transition Behaviour of Maltose-Water Mixtures . Carbohydrate Research , 212 : 109 – 117 .
- Peleg , M. 1992 . On the use of the WLF Model in Polymers and Foods . Critical Reviews in Food Science and Nutrition , 32 : 59 – 66 .
- Pezron , I. , Herning , T. , Djabourov , M. and Leblond , J. 1990 . “ Scattering From a Biopolymer Solution in the Sol and Gel States: the Gelatin Example ” . In Physical Networks, Polymers and Gels Edited by: Burchard , W. and Ross-Murphy , S. B. 231 – 252 . London : Elsevier .
- Richardson , R. K. and Kasapis , S. 1998 . “ Rheological Methods in the Characterisation of Food Biopolymers ” . In Instrumental Methods in Food and Beverage Analysis Edited by: Wetzel , D. L.B. and Charalambous , G. 1 – 48 . Amsterdam : Developments in Food Science, Volume 39, Elsevier .
- Roos , Y. 1993 . Melting and Glass Transitions of Low Molecular Weight Carbohydrates . Carbohydrate Research , 238 : 39 – 48 .
- Roos , Y. and Karel , M. 1991 . Plasticizing Effect of Water on Thermal Behavior and Crystallisation of Amorphous Food Models . Journal of Food Science , 56 : 38 – 43 .
- Rouse , P. E. Jr. 1953 . A Theory of the Linear Viscoelastic Properties of Dilute Solutions of Coiling Polymers . The Journal of Chemical Physics , 21 : 1272 – 1280 .
- Shen , M. C. and Eisenberg , A. 1967 . “ Glass Transitions in Polymers ” . In Progress in Solid State Chemistry Edited by: Reiss , H. 407 – 481 . Oxford : Pergamon Press .
- Slade , L. and Levine , H. 1991 . Beyond Water Activity: Recent Advances Based on an Alternative Approach to the Assessment of Food Quality and Safety . Critical Reviews in Food Science and Nutrition , 30 : 115 – 360 .
- Slade , L. and Levine , H. 1993 . “ The Glassy State Phenomenon in Food Molecules ” . In The Glassy State in Foods Edited by: Blanshard , J. M. and Lillford , P. J. 35 – 101 . Nottingham : Nottingham University Press .
- Sperling , L. H. 1986 . Introduction to Physical Polymer Science New York : Wiley-Interscience .
- Sworn , G. and Kasapis , S. 1998 . The use of Arrhenius and WLF Kinetics to Rationalise the Mechanical Spectrum in High Sugar Gellan Systems . Carbohydrate Research , 309 : 353 – 361 .
- orfield>Sworn , G. and Kasapis , S. 1999 . Molecular Origins of the Rheology of High-Sugar Gellan Systems . Progress in Colloid and Polymer Science , 114 : 116 – 122 .
- Thompson , A. B. and Woods , D. W. 1956 . The Transition of Polyethylene Terephthalate . Transactions of the Faraday Society , 52 : 1383 – 1397 .
- Tobolsky , A. V. 1956 . Stress Relaxation Studies of the Viscoelastic Properties of Polymers . Journal of Applied Physics , 27 : 673 – 685 .
- Tsoga , A. , Kasapis , S. and Richardson , R. K. 1999 . The Rubber-to-Glass Transition in high sugar agarose systems . Biopolymers , 49 : 267 – 275 .
- Ward , I. M. and Hadley , D. W. 1993 . “ Experimental Studies of Linear Viscoelastic Behaviour as a Function of Frequency and Temperature: Time-Temperature Equivalence ” . In An Introduction to the Mechanical Properties of Solid Polymers 84 – 108 . Chichester : John Wiley .
- Whorlow , R. W. 1992 . “ Dynamic Tests ” . In Rheological Techniques 275 – 333 . New York : Ellis Horwood .
- Williams , M. L. and Ferry , J. D. 1953 . Second Approximation Calculations of Mechanical and Electrical Relaxation and Retardation Distributions . Journal of Polymer Science , 11 : 169 – 175 .
- Williams , M. L. , Landel , R. F. and Ferry , J. D. 1955 . The Temperature Dependence of Relaxation Mechanisms in Amorphous Polymers and other Glass-Forming Liiquids . Journal of the American Chemical Society , 77 : 3701 – 3707 .
- Ziabicki , A. and Klonowski , W. 1975 . Topology, Thermodynamics and Physical Properties of Polymer Networks II. Thermodynamically Most Probable Structures and Related Physical Behaviour . Rheological Acta , 14 : 113 – 126 .