
Abstract
The protein arginine deiminase 4 (PAD4) is a calcium-dependent enzyme, which catalyses the irreversible conversion of peptidyl-arginines into peptidyl-citrullines and plays an important role in several diseases such as in the rheumatoid arthritis, multiple sclerosis, Alzheimer’s disease, Creutzfeldt–Jacob’s disease and cancer. In this study, we report the inhibition profiles and computational docking toward the PAD4 enzyme of a series of 1,2,3-triazole peptidomimetic-based derivatives incorporating the β-phenylalanine and guanidine scaffolds. Several effective, low micromolar PAD4 inhibitors are reported in this study.
Introduction
The protein arginine deiminases (PADs) are Ca2+-dependent enzymes responsible for the conversion of peptidyl-arginine residues to the corresponding citrulline derivatives through an irreversible hydrolytic post-translational process called citrullination (Scheme 1)Citation1.
Scheme 1. Conversion of peptidyl-arginine 1 into peptidyl-citrulline 2 catalyzed by PADsCitation1.
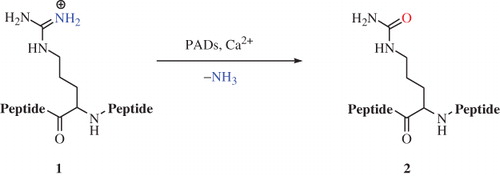
This reaction is involved in many biological events closely related to cellular growth and differentiationCitation1. Pathologies such as rheumatoid arthritisCitation2,Citation3, multiple sclerosis (MS)Citation4, Alzheimer’s diseaseCitation5,Citation6, Creutzfeldt–Jacob’s diseaseCitation7 and neutrophil extracellular trap formationCitation8,Citation9 are correlated to uncontrolled citrullination events operated by PADs. Therefore, there is an increasing attention for the development of new PAD ligands as potential drugs for the treatment of such pathologiesCitation10,Citation11.
PADs are present both in bacteria and mammalsCitation12,Citation13. In general, bacterial PADs (∼ 40 KDa) are composed only of the catalytic domain, whereas the mammalian counterparts (∼ 75 KDa) have the N-terminal domain further rearranged into two immunoglobulin-like sub-domainsCitation12,Citation13. Five distinct PAD isoforms were reported in humans (PAD1, 2, 3, 4 and 6), which differ for tissue and sub-cellular localizationsCitation14–18. PAD1 is mainly expressed in epidermisCitation14,Citation15, PAD2 in muscles and brainCitation16, PAD3 in hair folliclesCitation17,Citation18, PAD4 in granulocytesCitation19–21 and PAD6 is present in egg cells and embryosCitation22,Citation23. To date PAD4 is the only isoform easily accessible, being assumed to be a good model for all other isoformsCitation24.
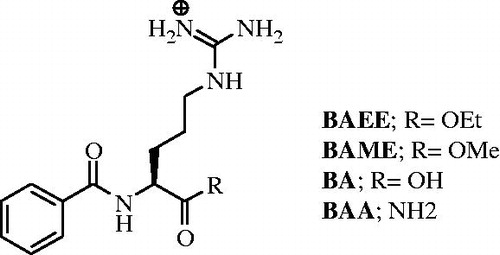
High resolution crystallographic investigations confirmed the dimeric nature of the PAD4 functional unit, which is composed of two monomers disposed in a head to tail arrangementCitation24. Each monomer is organized in two domains: the C- and the N-terminal domainCitation24. The first contains two highly specific sites for Ca2+, which appear highly conserved among the PAD enzymes except in the isoform 6Citation22,Citation24. Once in loco, the metal ions promote folding of the apoprotein to generate the active cleftCitation24. The presence of Ca2+ is also essential for the enzymatic activity, as demonstrated by kinetic and structural analyses confrontationsCitation24,Citation25. Additional mechanisms must be involved in the fine-tuning of the PAD4 activity, such as proteolytic splicings or protein–protein interactions, as the physiological intracellular concentrations of Ca2+ (∼ 1 μM in activated cells) are far below the calcium K0.5Footnotei reported when arginine derivatives such as BA, benzoyl-L-arginine amide (BAA), BAEE and BAME () are used as PAD substratesCitation26,Citation27.
Figure 1. Structures of the known PAD4-reversible inhibitors.
The remaining N-terminal subdomain, also referred as subdomain 1, is characterized by the presence of the nuclear localization signal (NLS) sequences, which are unique for PAD4Citation24. NLS sequences are responsible for the translocation of the protein from the cytoplasm into the cellular nucleus where it operates as transcriptional regulator through deimination of the H2A, H3 and H4 histonesCitation10,Citation11,Citation24. This feature makes PAD4 of particular relevance also for the potential development of new drugs useful for the treatment of pathologies where alterations of the genetic expression are implicated as in the cancerCitation28,Citation29 and, as recently demonstrated, also in the MSCitation30.
The proposed citrullination mechanism of PAD4 is well reportedCitation31 and accounts for the substrate containing the guanidine group, being correctly allocated and hold in place into the enzymatic cavity by means of a hydrogen bond network involving two acidic residues, Asp350 and Asp473Citation31. A nucleophilic attack of the highly conserved Cys645 residue generates a tetrahedral intermediate, which collapses with the assistance of the His471, to afford the S-thioalkyluronium derivative and ammonia. The latest step includes the alkaline hydrolysis of the S-thioalkyluronium species and the release of the citrullinated substrateCitation28,Citation31. The generation of the hydroxide species required for the hydrolytic step necessitates the His471 residue to be correctly placed and orientated with respect to the progenitor water molecule. However in PAD4, such a residue does not have the adequate surrounding amino acids to ensure the deprotonationCitation31. A product-assisted catalysis mechanism has been proposed with the generated ammonia participating in the formation of the hydroxide speciesCitation31. However, experimental data are also in agreement with an alternative mechanistic theory where the substrate itself might play an important role in ensuring the generation of the hydroxide ion in PAD4Citation32.
The identification of small molecules as PAD substrates (PADSs) and having an acceptable degree of selectivity among the various isoforms represents a key step for the development of new lead compounds useful for the treatment of pathologiesCitation33.
Materials and methods
Chemistry
Anhydrous solvents and all reagents were purchased from Sigma-Aldrich, Alfa Aesar and TCI (Milan, Italy). All reactions involving air- or moisture-sensitive compounds were performed under a nitrogen atmosphere using dried glassware and syringes techniques to transfer solutions. Nuclear magnetic resonance (1H-NMR, Citation13C-NMR and APT) spectra were recorded using a 200 MHz Varian Gemini spectrometer in CDCl3, MeOH-d4 or DMSO-d6 (Turin, Italy). Chemical shifts are reported in parts per million, and the coupling constants (J) are expressed in Hertz (Hz). Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet; br, broad singlet; and dd, double of doublets. The assignment of exchangeable protons (OH and NH) was confirmed by the addition of D2O. Analytical thin-layer chromatography was carried out on Merck silica gel F-254 plates. Flash column chromatography purifications were performed on Merck Silica gel 60 (230–400 mesh ASTM) as the stationary phase and ethylacetate/n-hexane or MeOH/DCM were used as eluents. ESI mass spectra were carried out on a ion-trap double quadrupole mass spectrometer using electrospray (ES+) ionization techniques and a normalized collision energy within the range of 25–32 eV for MSMS experiments. Optical rotations were determined on a JASCO DIP-370 instrument.
General procedure for the CuAAC synthesis of compounds 1–16
A stirred solution of alkyne (1.0 eq) and azide (1.0 eq) in H2O/t-BuOH 1/1 (4 mL/mmol) was treated under a nitrogen atmosphere with a sodium ascorbate 0.9 M solution (1.0 eq) and a Cu(OAc)2 0.3 M solution (1.0 eq)Citation34. The mixture was stirred at r.t. for two days. Then the organic phase was extracted with DCM (× 3), washed with a 5% solution of NaHCO3 (× 3) and brine (× 2), dried over Na2SO4 and evaporated under vacuo to give a residue that was purified by silica gel column chromatography to afford the pure protected product. The title compounds were obtained as hydrochloride salts by acid catalyzed hydrolysis at r.t. for 16 h with 3 M HCl solution (5 mL/mmol).
General procedure for the synthesis of guanidine-containing alkynes
To a solution of alkyn-1-ol (1.0 eq) in dry THF were added, under nitrogen, PPh3 (1.0 eq) and N,N′-di-Boc-guanidine (1.0 eq)Citation34. DIAD (1.0 eq) was slowly added at 0 °C, then the solution was heated at 110 °C in microwave for 30 min. The crude mixture was purified through silica gel column chromatography eluting with EtOAc/petroleum ether 1/2 to afford the pure product.
Enzymatic assay
hPAD4 enzyme is from Cayman (item no. 10500). Antibody-based assay for PAD activity (ABAP) kit from Modiquest (Cat.-no. MQ-17.101). Deimination buffer: 40 mM Tris-HCl, 5 mM CaCl2, 1 mM DTT, pH 7.5 wash buffer: PBS w. 0.05% Tween-20.
Modified protocol from Modiquest
Preincubate 96-well plate for 30 min at 37 °C with 100 µl deimination bufferCitation35. Preincubate PAD4 enzyme with compound (final concentration and 0.1% DMSO) in deimination buffer for 15 min at 37 °C. Empty the ABAP plate by inversion. Transfer 100 µl hPAD4/compound solution to the ABAP plate (100 µl total volume/well). Incubate the ABAP plate for 20 min at 37 °C in a humidified chamber. Wash the plate five times with washing buffer. Dilute the MQR mouse anti-deiminated antibody 1:1000 in washing buffer + 1% BSA. Add 100 µl antibody solution to each well. Incubate for one hour at 37 °C in a humidified chamber. Wash the plate five times with wash buffer. Dilute the HRP-labeled anti-mouse Ig antibody 1:2000 in wash buffer + 1% BSA. Add 100 µl antibody solution to each well. Incubate for one hour at 37 °C in a humidified chamber. Wash the plate five times with wash buffer. Wash the plate three times with wash buffer without Tween-20. Add 100 µl TMB substrate in each well and let the blue color develop (room temperature). Add 100 µl 2 M H2SO4 in each well to stop the reaction. Read OD at 450 nm in a multi-well plate reader.
Molecular modeling
X-ray crystallographic complex of PAD4 with BAA was retrieved from PDB Data Bank (http://www.rcsb.org/ – PDB code: 1WDA). Since some catalytically irrelevant residues are missed, the N- and C-termini of the resulting gaps were capped with acetyl groups and N-methyl, respectively. Moreover, being 1WDA the C645A mutant of WT PAD4, a reverse mutation of Ala to Cys in position 645 was performed. Thus capped residues and Cys645 were minimized using AMBER99 force field. Again, missing hydrogens were added, and partial charges and protonation state were assigned with a generalized Born solvation model using previously selected AMBER99 force field. The database of the three-dimensional structures of evaluated compounds were built in Molecular Operating Environment (MOE), the protonation state of each structure at neutral pH was automatically calculated, then partial charges and geometrical optimization and minimization using the modified MMFF94x force field was performed using a generalized Born solvation model and RMSD gradient 0.05 kcal mol−1 Å−1.
Rigid receptor-flexible ligand docking calculations were performed using the docking simulation feature MOE-dock by setting grid sizes that included the entire macromolecules. The triangle matcher was used as placement method to generate docking posesCitation36, and the London ΔGbinding scoring function that estimates the free energy of binding expressed in kcal/mol, was used to rank hit candidates, after a force field-based refinement and rescoring.
Results and discussion
Chemistry
In this study, we investigate a series of peptidomimetic-based derivatives 1–16 (Schemes 2–4), incorporating the primary guanidine group placed at the terminal-end, as reversible PAD4 inhibitors.
With the exception of 4, 5, 8 and 10, which are in this study reported for the first time, all compounds of the series were identified by some of us as potential antiangiogenic agentsCitation37. In this study, as extension of our previous study on PAD4 inhibitorsCitation37, we evaluate compounds 1–16 as reversible PAD4 inhibitors. The main feature of such derivatives is the presence of the guanidine moiety mimicking the same group in the PAD substrates.
All compounds were obtained via the 1,3-Huisgen cycloaddition reaction using stoichiometric amounts of the Cu(II) acetate salt for the combination of terminal azides (Intermediate A for 1–15 and Intermediate A1 for 16) and alkynes to afford the final 1,2,3-triazoles derivatives 1–16 upon acidic cleavage of both protecting groups at the terminal ends (Schemes 2–4)Citation34.
The alkyl–azide intermediates A were synthesized by reaction of the appropriate β-amino ester moieties with bromoacetyl bromide, followed by displacement of the alkylbromide with sodium azide (Scheme 2A)Citation34. The terminal alkyne fragments were obtained either via the Mitsunobu reaction coupling of the appropriate alkyn-1-ols with di-Boc-guanidine (Scheme 2B), or by nucleophilic substitution on the commercially available di-Boc-guanidine triflate with the appropriate alkynyl piperazines (Scheme 3).
Scheme 2. Reagents and conditions: (i) Ph3P, DIAD and THF; (ii) Cu(OAc)2, Na ascorbate, H2O/t-BuOH 1/1 and intermediate A; and (iii) HClaq 3M [Citation34].
![Scheme 2. Reagents and conditions: (i) Ph3P, DIAD and THF; (ii) Cu(OAc)2, Na ascorbate, H2O/t-BuOH 1/1 and intermediate A; and (iii) HClaq 3M [Citation34].](/cms/asset/4f3bd44e-1d3c-4000-82f1-4c4369d9c0f4/ienz_a_947976_sch0002_c.jpg)
Scheme 3. Reagents and conditions: (i) Et3N and DCM; (ii) Cu(OAc)2, Na ascorbate, H2O/t-BuOH 1/1, Intermediate A; and (iii) HClaq 3M [Citation34].
![Scheme 3. Reagents and conditions: (i) Et3N and DCM; (ii) Cu(OAc)2, Na ascorbate, H2O/t-BuOH 1/1, Intermediate A; and (iii) HClaq 3M [Citation34].](/cms/asset/5fb17603-99bc-412d-840b-cbda588b4d26/ienz_a_947976_sch0003_c.jpg)
A similar approach was employed for the synthesis of compound 16 using the N-phenylsubstituted glycine as starting intermediate (Scheme 4)Citation34.
Scheme 4. General synthetic scheme of compound 16 [Citation34].
![Scheme 4. General synthetic scheme of compound 16 [Citation34].](/cms/asset/e235b3bb-619b-4666-a0a1-0b2b96aa4396/ienz_a_947976_sch0004_c.jpg)
Among the enzymatic assays reported for the evaluation of compounds inhibitory activities toward the PAD4 enzymeCitation35–39, we considered a commercially available antibody-based kitFootnoteii. Such an assay determines the conversion of the arginine peptide residues to the corresponding citrullines operated by the PAD4 by means of antibodies (details are reported in the experimental section). The main advantage of such a method mainly relays on the small amount of PAD4 enzyme required. Screenings were carried out at two different concentrations (1 μM and 10 μM), and all results, expressed as inhibition percentages, are the mean of three different runs. Chloroamidine as standard PAD4 irreversible inhibitor has also been included in the assay ().
Table 1. ABAP assay results at 1 μM and 10 μM concentration of inhibitora.
All compounds showed inhibition properties against PAD4 at both concentrations with percentage values comprised between 5 and 41% at 1 μM, with the exception of 2, 4, 7, 11, 13, which are inactive, and between 16 and 99% at 10 μM.
We identified the enantiomeric nature at the β-amino acidic portion and the spacer between the carboxylic acid and the terminal guanidine functionalities as the main factors influencing the PAD4 inhibitory activities. Compound 1, the shortest in the series in racemic form, showed a 7% inhibition at 1 μM and 76% at 10 μM, whereas its (S)-enantiomer 3 registered a significant increase at the lower concentration (19%) and the (R)-enantiomer 2 only moderately inhibits at 10 μM (37%). Interestingly, the elongation of one carbon unit at the α-carbonyl position in 1 to give the racemic 11 suppressed the inhibitory activity at 1 μM and halved at 10 μM (36%). Insertion of the same spacer between the triazole and the guanidine moieties, as for compound (S)-9, restored the inhibition properties (26% at 1 μM and 69% at 10 μM), which were further enhanced when the stereogenic center was reversed and a fluorine atom was introduced as in compound 8 (39% inhibition at 1 μM and 99% at 10 μM). Additional elongation of one carbon unit as for compounds (S)-7 and (R)-6 resulted in a marked dropping of the inhibition (0% at 1 μM, 53% at 10 μM and 18% at 1 μM, 82% at 10 μM, respectively). Interestingly, the introduction of a fluorine atom as in (R)-4 and in (S)-5 suppressed the inhibitory activities at 1 μM (0% and 5%, respectively) but maintained good potencies at 10 μM (76% and 70%, respectively). In this case, no substantial differences related to the enantiomeric nature of the compounds were observed. Moreover, we explored the effects of removal of the phenyl group at the β-amino acidic portion but keeping the three-carbon unit spacer, as for the compound 10. Such modifications did not significantly improve the inhibition (19% at 1 μM and 73% at 10 μM).
Analogous SAR evaluations were carried out when a piperazine group was present between the guanidine and the triazole moieties. Compounds (R)-12 and (S)-13 showed from low to medium inhibitory values (26% at 1 μM, 16% at 10 μM for (R)-12 and 0% at 1 μM, 20% at 10 μM for (S)-13). A sensible enhancement was observed for the one carbon unit longer derivatives (R)-14 and (S)-15 (41% 1 μM, 99% at 10 μM for (R)-14 and 14 at 1 μM, 72% at 10 μM for (S)-15).
Interestingly, the α-amino acid glycine derivative 16 showed inhibition values comparable with the β-analogues (18% at 1 μM and 99% at 10 μM).
In summary, a rather complex SAR analysis can be depicted. Most of the compounds in this study reported showed good inhibition profiles on the PAD4 enzyme. Elongation of the chain between the guanidine and the 1,2,3-triazole moieties resulted in significant changes in the inhibition potencies. However, the interconversion of the stereogenic centers at the aminoacidic portions as well as the introduction of a halogen atom at the phenyl ring heavily affect the kinetic results. Among all the series, herein reported compounds 8, 14 and 16 are the most powerful in inhibiting the PAD4 enzyme in vitro.
Molecular modeling
In order to assess a reasonable binding mode of the compounds herein proposed with the PAD4 enzyme, we docked their three-dimensional structures into the optimized PAD4 crystallographic structure (PDB code: 1WDA), and the coherent best-ranked pose of each compound was retained and examined with the MOE program (supporting informations)Footnoteiii.
Interestingly, all proposed compounds shared similar binding modes within the PAD4 enzymatic cleft. In particular, the guanidine moieties were deeply buried within the active site and are held in place by a hydrogen bond network with the Asp350 and Asp473 residues ().
Figure 2. In silico docking of title compounds within the binding site of PAD4 protein structure. Top-ranked energy poses and representative view of the C-terminal domain. The binding site is represented as molecular surfaces, and ligands and key amino acid residues are shown as pink and green sticks, respectively.
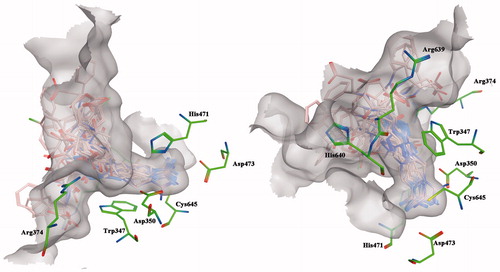
Further stabilizations are ensured by the interactions occurring between the internal amides and/or the external carboxylic moieties of the title compounds with the basic aminoacid residues Arg374, Arg639 and His640 (). Interestingly, the common 1,2,3-triazole moieties interact via hydrogen bondings with the Arg374 residue or via π-interactions with the Trp347 depending on the side chain lengths ().
Figure 3. (A) Overall view of binding modes of the 1,2,3-triazoles and guanidinium moieties on compounds 1–16. Detailed views of model compounds 6 (B) and 3 (C).
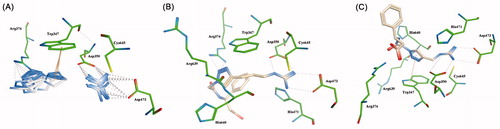
Such a binding feature accounts for additional stabilization of the compound-PAD4 enzyme complexes. The ability of the 1,2,3-triazole group to switch from hydrogen bond to π-interactions with the proper amino acid residues depending on the side chain lengths has some reflections on the inhibition potencies as previously reported. We consider such a structural feature a key motif for designing peptidomimetic-based molecules as PAD4-reversible inhibitors and additional work is currently ongoing to better clarify this point.
In summary, we report a series of peptidomimetic-based compounds 1–16, which showed appreciable inhibition profiles for the PAD4 enzyme in vitro at two different concentrations, and their data were compared with the commercially available and irreversible inhibitor chloroamidine.
The putative binding modes of 1–16 were also investigated by means of molecular modeling calculations, which revealed the 1,2,3-triazole ring having specific binding features with the Arg374 or with the Trp347 residue depending on the side chain lengths.
Therefore, we consider the use of peptidomimetics as a promising approach for the development of new and reversible inhibitors of the PAD4 enzyme. Moreover, the development of non-covalent PAD inhibitors indeed represents a valuable opportunity for the selective targeting among the different isoforms, and hence reduces the multitude of side effects deriving from aspecific interactions.
Supplemental Material.pdf
Download PDF (138.6 KB)Acknowledgements
The authors thank Dr. Andrea Brancale for his assistance on the elaboration of the molecular modeling calculation.
Declaration of interest
The authors confirm that this article content has no conflict of interest.
I. K., T. D., A. S., C. T. S. and F. C. are grateful to an EU FP7 project, Gums & Joints, for financial support (Grant agreement: HEALTH-F2-2010-261460). M. S. and N. P. gratefully acknowledge the “Fondazione Banco di Sardegna” for its partial financial support.
Notes
iK05 is defined as the concentration of calcium corresponding to half of the maximalmenzyme activity.
iiiThe antibody-based kit for PAD4 activity measurements was obtained from Modiquest Research, AJ Nijmegen, The Netherlands (Cat. no. MQ-17.101-96) and provides a 96-well microplate in which the bottom of each reaction well is coated with arginine containing peptides as the substrate for PAD4.
iiiMolecular Operating Environment (MOE 2009.10). Chemical Computing Group, Inc. Montreal, Quebec, Canada.
References
- Shirai H, Blundell TL, Mizuguchi K. A novel superfamily of enzymes that catalyze the modification of guanidino groups. Trends Biochem Sci 2001;26:465–8
- Wegner N, Lundberg K, Kinloch A, et al. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol Rev 2010;233:34–54
- Suzuki A, Yamada R, Yamamoto K. Citrullination by peptidylarginine deiminase in rheumatoid arthritis. Ann NY Acad Sci 2007;1108:323–39
- Wood DD, Bilbao JM, O’Connors P, Moscarello MA. Acute multiple sclerosis (Marburg type) is associated with developmentally immature myelin basic protein. Ann Neurolog 1996;40:18–24
- Ishigami A, Ohsawa T, Hiratsuka M, et al. Abnormal accumulation of citrullinated proteins catalyzed by peptidylarginine deiminase in hippocampal extracts from patients with Alzheimer's disease. J Neurosci Res 2005;1:120–8
- Mohlake P, Whiteley CG. Arginine metabolising enzymes as therapeutic tools for Alzheimer's disease: peptidyl arginine deiminase catalyses fibrillogenesis of beta-amyloid peptides. Mol Neurobiol 2010;41:149–58
- Jang B, Jin JK, Jeong YC, et al. Involvement of peptidylarginine deiminase-mediated post-translational citrullination in pathogenesis of sporadic Creutzfeldt-Jakob disease. Annals Neuropathol 2009;119:199–210
- Knight JS, Zhao W, Luo W, et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest 2013;7:2981–93
- Rohrbach AS, Slade DJ, Thompson PR, Mowen KA. Activation of PAD4 in NET formation. Front Immunol 2012;3:360. doi: 10.3389/fimmu.2012.00360
- Sunish M, Cherrington BD, Horibata S, et al. Potential role of peptidylarginine deiminase enzymes and protein citrullination in cancer pathogenesis. Biochem Res Int 2012;2012:895343. doi: 10.1155/2012/895343
- Bicker KL, Thompson PR. The protein arginine deiminases: structure, function, inhibition, and disease. Biopolymers 2013;2:155–63
- Shirai H, Blundell TL, Mizuguchi K. A novel superfamily of enzymes that catalyze the modification of guanidino groups. Trends Biochem Sci 2001;26:465–8
- Shirai H, Mokrab Y, Mizuguchi, K. The guanidino-group modifying enzymes: structural basis for their diversity and commonality. Proteins 2006;4:1010–23
- Senshu T, Aliyama K, Ishigami A, Nomura K. Studies on specificity of peptidylarginine deiminase reactions using an immunochemical probe that recognizes an enzymatically deiminated partial sequence of mouse keratin K1. J Dermatol Sci 1999;21:113–26
- Yamamoto I, Senshu T, Eady RA, et al. Sequential reorganization of cornified cell keratin filaments involving filaggrin-mediated compaction and keratin 1 deimination. J Invest Dermatol 2002;118:282–7
- Moscarello MA, Pritzker LB, Mastronardi FG, Wood DD. Peptidylarginine deiminase: a candidate factor in demyelinating disease. J Neurochem 2002;81:335–43
- Kanno T, Kawada A, Yamanouchi J, et al. Human peptidylarginine deiminase type III: molecular cloning and nucleotide sequence of the cDNA, properties of the recombinant enzyme, and immunohistochemical localization in human skin. J Invest Dermatol 2000;115:813–23
- Rogers G, Winter B, McLaughlan C, et al. Peptidylarginine deiminase of the hair follicle: characterization, localization, and function in keratinizing tissues. J Invest Dermatol 1997;108:700–7
- Nakashima K, Hagiwara T, Ishigami A, et al. Molecular characterization of peptidylarginine deiminase in HL-60 cells induced by retinoic acid and 1alpha,25-dihydroxyvitamin D(3). J Biol Chem 1999;274:27786–92
- Hagiwara T, Nakashima K, Hirano H, et al. Deimination of arginine residues in nucleophosmin/B23 and histones in HL-60 granulocytes. Biochem Biophys Res Commun 2002;290:979–83
- Nakashima K, Hagiwara T, Yamada M. Nuclear localization of peptidylarginine deiminase V and histone deimination in granulocytes. J Biol Chem 2002;277:49562–8
- Chavanas S, Méchin MC, Takahara H, et al. Comparative analysis of the mouse and human peptidylarginine deiminase gene clusters reveals highly conserved non-coding segments and a new human gene, PADI6. Gene 2004;330:19–27
- Wright PW, Bolling LC, Calvert ME, et al. ePAD, an oocyte and early embryo-abundant peptidylarginine deiminase-like protein that localizes to egg cytoplasmic sheets. Dev Biol 2003;256:73–88
- Arita K, Hashimoto H, Shimizu T, et al. Structural basis for Ca(2+)-induced activation of human PAD4. Nat Struct Biol 2004;11:777–83
- Wonhwa C. Membrane targeting by C1 and C2 domains. J Biol Chem 2001;276:32407–10
- Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol 2001;19:497–521
- Takahara H, Tsuchida M, Kusubata M, et al. Peptidylarginine deiminase of the mouse. Distribution, properties, and immunocytochemical localization. J Biol Chem 1989;264:13361–8
- Thompson PR, Fast W. Histone citrullination by protein arginine deiminase: is arginine methylation a green light or a roadblock? ACS Chem Biol 2006;7:433–41
- Slack JL, Causey CP, Thompson PR. Protein arginine deiminase 4: a target for an epigenetic cancer therapy. Cell Mol Life Sci 2011;68:709–20
- Lianhu W, Wasilewski E, Kumar Chakka S, et al. Novel inhibitors of protein arginine deiminase with potential activity in multiple sclerosis animal model. J Med Chem 2013;56:1715–22
- Kearney PL, Bhatia M, Jones NG, et al. Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 2005;44:10570–82
- Galkin A, Lu X, Dunaway-Mariano D, Herzberg O. Michaelis complex and the thiouronium reaction intermediate of Pseudomonas aeruginosa arginine deiminase. J Biol Chem 2005;280:34080–7
- Dreyton CJ, Anderson ED, Subramanian V, et al. Insights into the mechanism of streptonigrin-induced protein arginine deiminase inactivation. Insights into the mechanism of streptonigrin-induced protein arginine deiminase inactivation. Bioorg Med Chem 2014;22:1362–9
- Trabocchi A, Menchi G, Cini N, et al. Click-chemistry-derived triazole ligands of arginine-glycine-aspartate (RGD) integrins with a broad capacity to inhibit adhesion of melanoma cells and both in vitro and in vivo angiogenesis. J Med Chem 2010;53:7119–28
- Bozdag M, Dreker T, Henry C, et al. Novel small molecule protein arginine deiminase 4 (PAD4) inhibitors. Bioorg Med Chem Lett 2013;23:715–19
- Yuki H, Ohmura T, Kawasaki H, et al. Clinical analysis of urea nitrogen: chemical structure of the colored reaction product of urea with diacetylmonoxime and thiosemicarbazide. Chem Pharm Bull 1981;29:1096–100
- Sugawara K, Oyama O. Fluorogenic reaction and specific microdetermination of ammonia. J Biochem 1981;89:771–4
- Sternson LA, Stobaugh JF, Repta A. Rational design and evaluation of improved o-phthalaldehyde-like fluorogenic reagents. J Anal Biochem 1985;144:233–46
- Mana H. Rapid and selective determination of ammonium by fluorimetric flow injection analysis. [PhD Thesis]. Martin-Luther-Universität Halle-Wittenberg; November 1998
Supplementary material available online Supporting information