
Abstract
Signal transducer and activator of transcription 3 (STAT3) plays an essential role in cell growth regulation and survival. An aberrant STAT3 activation and/or expression is implied in various solid and blood tumors as well as in other pathologies like rheumatoid arthritis and pulmonary fibrosis, thus making the search for STAT3 inhibitors a growing field of study. With the aim of identifying new inhibitors of STAT3 dimerization, we screened a database including more than 1 320 000 commercially available compounds using a receptor-based pharmacophore model comprising the key protein–protein interactions identified in the STAT3 dimer and refining the search through docking and molecular dynamic simulations studies. STAT3 binding assays revealed a significant STAT3 inhibitory activity and selectivity versus Grb2 for one of the four top-scored compounds, thus verifying the reliability of the virtual screening workflow. Moreover, such compound could already be considered as a lead for the development of new and more potent STAT3 dimerization inhibitors.
Introduction
Signal transducer and activator of transcription 3 (STAT3) is a member of the STAT family, made up of latent cytosolic proteins which directly mediate signals for the plasma membrane (in response to cytokines and growth factors) to the nucleusCitation1 playing a key role in cell growth regulation and survivalCitation2. STAT3 contains seven structurally and functionally conserved domains among which the Src homology 2 (SH2) domain is essential for its activation cascade pathway. Indeed, STAT3 monomer phosphorylation at Tyr705 residue, by tyrosine kinases (Janus kinases, tyrosine protein kinase 2 and c-Src kinases), leads to STAT3–STAT3 dimerization, through reciprocal pTyr–SH2 domain interaction. The dimer complex translocates into the nucleus and, binding to specific DNA consensus sequences, induces target gene transcriptionCitation2. Nevertheless, due to the de-regulation of cytokine receptors, growth factors and Janus kinases activityCitation3, STAT3 turns out to be constitutively activated in a wide variety of human solid and blood tumors, involving uncontrolled growth and survival of cells, enhanced angiogenesis and metastasisCitation4,Citation5. Noteworthy, apoptosis was induced inhibiting STAT3 only in cancer cell lines, with slight effect in normal cellsCitation6.
Therefore, due to the crucial role of an aberrant STAT3 expression and/or activation in cancer development as well as its implications in other pathologies, like rheumatoid arthritis, atherosclerosis, inflammatory bowel disease, psoriasis and pulmonary fibrosisCitation7, the search for STAT3 inhibitors is a hot topic in medicinal chemistry. A considerable number of different types of direct small molecule STAT3 inhibitors, preventing STAT3 dimerization or STAT3–DNA binding and comprising peptides, peptidomimetics and oligonucleotides, has been discoveredCitation6–8. Nevertheless, the development of highly potent and selective STAT3 inhibitors is still a challenging task. Computer screening proved to be effective in the identification of new STAT3 SH2 domain binding inhibitors, blocking STAT3 dimerization, including some well-known compounds like STA-21Citation9, S3I-201Citation10 and STX-0119Citation11, which have been all identified through in silico screening purely relied on the docking of compound libraries into the STAT3 SH2 domain. Other examples of docking-based virtual screening (VS) studies are reported in literatureCitation12–15 and recently some fragment-based drug design studies, combining docking and synthesis, have been publishedCitation16,Citation17. However, different computational approaches, like QSAR studies and pharmacophore screening, have been rarely appliedCitation18,Citation19. In this study, we report one of the first VS studies relied on a receptor-based pharmacophore model comprising the key protein–protein interactions identified in the STAT3 dimer. The model has been used to screen a database of commercially available compounds, thus selecting small molecules endowed with the features necessary to bind the STAT3 SH2 domain. Then, a docking approach and molecular dynamic (MD) simulations were used to refine the screening and to identify new potential inhibitors of STAT3 dimerization ().
Figure 1. Scheme of the VS workflow.
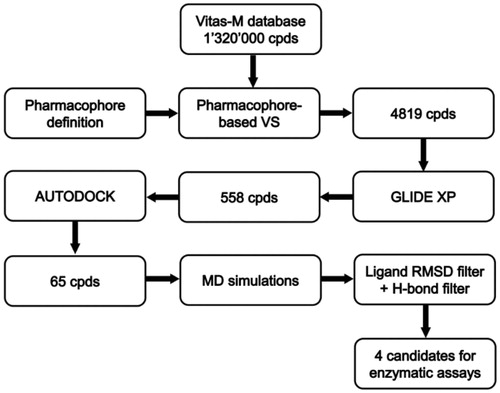
Materials and methods
Receptor-based pharmacophore model generation
The receptor-based pharmacophore model was built based on the 3D structure of the STAT3β homodimer bound to DNA. The X-ray structure of the STAT3β co-crystallized with a DNA fragment (PDB code 1BG1)Citation20 was downloaded from the Protein Data BankCitation21 and the STAT3β homodimer was built by applying the transformation matrix as reported in the PDB file with the removal of the DNA fragment. For the generation of the pharmacophore model, the sequence corresponding to residues M586-F716 of one of the STAT3β monomers, comprising its SH2 domain, was taken into account as the receptor while the phosphopeptide AAPpYLKT belonging to the other monomer (residues A702-T708) was considered as the ligand. The receptor-based pharmacophore model was created with the software PHASECitation22 by automatically generating an exhaustive model comprising all the possible pharmacophoric features identified by the program for the AAPpYLKT peptide. Then, only the four features representing the key protein–protein interactions established by the residues pTyr705 and Leu706 of the phosphopeptide were considered in the final pharmacophore model, while the other ones were removed.
The excluded volume of the receptor was created using the create_xvolReceptor utility of PHASE. The excluded volume spheres, representing regions of space that cannot be occupied by the compounds when aligned to the model during a pharmacophore search, were created taking into account the receptor atoms at a distance comprised between 2 and 5 Å from the ligand surface, while leaving all other settings as their defaults.
Database generation and pharmacophore screening
The Vitas-M database, comprising about 1 320 000 commercially available compounds was used as the screening database. The phasedb_manage and phasedb_confsites utilities of PHASECitation22 were used to create a PHASE 3D database in which conformational ensembles and pharmacophore sites were created and stored for each compound. The conformational sampling method was set to “thorough”, so that a complete set of conformations was generated for both the core and the peripheral groups of the compounds, while all other settings where left as their defaults.
The PHASE 3D database was then screened using the previously created receptor-based pharmacophore model and the receptor excluded volume, imposing that only the compounds matching all the four pharmacophoric features of the model were retrieved.
Docking procedures and pose filtering
In all the docking procedures, the compounds were docked into the crystal structure of the STAT3β co-crystallized with DNA (PDB code 1BG1)Citation20. Prior to docking, the DNA fragment was removed and only the sequence of one STAT3β monomer corresponding to residues M586-F716, comprising its SH2 domain, was used as receptor for the calculations. The top-scored poses of the docked ligands were taken into account as a result of each docking procedure.
GLIDE 5.0
The docking region was defined by a parallelepiped box of 30 Å in the x direction and 25 Å in the y and z directions that was centered on the bound phosphopeptide AAPpYLKT. The option allowing only the docking of ligands containing a defined range of atoms was disabled, so that all the ligands were docked independently from their number of atoms. The extra precision (XP) method was used for the study and all other settings were left as GLIDECitation23 defaults.
AUTODOCK 4.2.3
AUTODOCK ToolsCitation24 was used to define the torsion angles in the ligands, to add the solvent model and to assign partial atomic charges to the ligands and the protein. The docking site used for AUTODOCKCitation25 calculations was defined considering the bound phosphopeptide AAPpYLKT as the central group of a grid of 80, 70 and 70 points in the x, y and z directions. The energetic maps were calculated using a grid spacing of 0.375 Å and a distance-dependent function of the dielectric constant. The ligands were subjected to 200 docking runs of the AUTODOCK search using the Lamarckian genetic algorithm (LGA) and employing 10 000 000 energy evaluations; the number of individuals in the initial population was set to 500 and a maximum of 10 000 000 generations were simulated during each docking run; an rms tolerance of 1.5 Å was used to carry out the cluster analysis of the docking solutions and all the other settings were left as their defaults.
Filtering of docking results
The filtering of the docking results was carried out by superimposing the top-scored poses of the docked compounds to the pharmacophore model through a PHASE pharmacophore search in which the “scoreInPlace” option was enabled, so that the matching to the model was computed directly from the supplied poses, without changing their coordinates. The retrieval of compounds matching all the four pharmacophoric features of the model was imposed in this search.
MD simulations
All simulations were carried out using AMBER 11Citation26. The simulation protocol was set up using the complex of the phosphopeptide AAPpYLKT bound to the SH2 domain of STAT3 as a reference. Prior to the MD simulations, the missing loop portion P689-S701 was automatically constructed within the structure of the STAT3β monomer (PDB code 1BG1) and then refined using the loop optimization method of the Modeller softwareCitation27. Only the sequence of STAT3 corresponding to residues M586-F716, comprising the SH2 domain, was taken into account as the receptor for the MD simulations of both the reference complex and the ligand–protein complexes of the compounds selected through the docking studies. The complexes were placed in a parallelepiped water-box (TIP3P explicit solvent model) and solvated with a 10 Å water cap. Sodium ions were added as counterions for the neutralization of the system. General amber force field (GAFF) parameters were assigned to the ligands, while partial charges were calculated using the AM1-BCC method. Before running the MD simulations, two steps of minimization were carried out. In the first step, a position restraint of 100 kcal/(mol ċ Å2) was applied to the complex, so that only the position of the water molecules was minimized. In the second step, a harmonic potential of 10 kcal/(mol ċ Å2) was applied only to the protein α carbons; thus, the whole system was energy minimized through 10 000 steps of steepest descent followed by conjugate gradient, until the attainment of a convergence of 0.05 kcal/(mol ċ Å2). The energy minimized complexes were employed as the starting structures for the MD simulations, which were run using particle mesh Ewald electrostatics and periodic boundary conditionsCitation28, a cutoff of 10 Å for the non-bonded interactions and employing SHAKE algorithm to keep rigid every bond involving hydrogen. For each complex, a 2 ns MD simulation, with a time step of 2.0 fs, was carried out. For the first 300 ps of simulation, constant-volume periodic boundary conditions were used and the temperature was raised from 0 to 300 K; then, constant-pressure periodic boundary conditions were employed for the remaining 1.7 ns of MD simulation, during which the temperature of the system was kept constant at 300 K using the Langevin thermostat. The analysis of the MD trajectories to calculate the RMSD of the ligands respect to their starting position, as well as the percentage of occupancy of their H-bonds were performed using the Ptraj suite of AMBER 11.
Alpha-screen based assays
STAT3 inhibitory activity of the commercially available compounds VS1–4 was tested by the AlphaScreen-based assay to evaluate the potential inhibition of the interaction between STAT3 SH2 domain and pTyr-containing peptides at 30 μM concentration, using MD77Citation29 as reference compound. For the most interesting compound, VS1, selectivity tests versus STAT1 and Grb2 (Growth factor receptor-bound protein 2) were performed. AlphaScreen is a bead-based non-radioactive assay system for detecting biomolecular interactions in a microtiter plate formatCitation30. Binding of biological partners brings donor and acceptor beads into close proximity and as result, a fluorescent signal between 520 and 620 nm is produced. The AlphaScreen-based assays were performed in a final reaction volume of 25 μL of the assay buffer containing 10 mM HEPES–NaOH (pH 7.4), 50 mM NaCl, 1 mM EDTA (pH 8.0), 0.1% NP-40 and 10 ng/μL BSA in a 96-well microtiter plate at 25°C. Phospho-Tyr (pTyr) peptide probes used in this study were 5-carboxyfluorescein (FITC)-GpYLPQTV for STAT3, FITC-GpYDKPHVLfor STAT1 and FITC-PSpYVNVQN for Grb2. First, 75 nM of each SH2-containing protein was incubated with the test compound for 15 min. Each protein sample was then incubated for 90 min with 50 nM of its corresponding FITC-pTyr peptide, and mixed with streptavidin coated donor beads and anti-FITC acceptor beads simultaneously before detection at 570 nm using EnVisonXcite (Perkin Elmer, Waltham, MA).
Results and discussion
With the aim of identifying novel STAT3 SH2 inhibitors, we developed a VS protocol focused on a receptor-based pharmacophore model comprising the key interactions established between the SH2 domains of the two STAT3 monomers within the STAT3–STAT3 dimer. The pharmacophore model was created based on the analysis of the crystal structure of the STAT3β homodimer co-crystallized with a DNA fragment (PDB code 1BG1)Citation20 (see “Materials and methods” section for details). shows the phosphopeptide AAPpYLKT, corresponding to residues 702–708 of one of the STAT3β monomer, bound to the SH2 domain of the other monomer. The central residues pTyr705 and Leu706 seem to be responsible of the major interactions formed by the peptide; in fact, alanine scanning mutagenesis studies revealed that these two residues are essential for the binding to the STAT3 SH2 domain, since tripeptides containing the pTyr-Leu portion (like Ala-pTyr-Leu or Pro-pTyr-Leu) were sufficient to significantly inhibit STAT3 DNA-binding activityCitation31.
Figure 2. Structure of the phopshopeptide AAPpYLKT (stick), bound to the STAT3 SH2 domain (shown as protein surface) obtained from the structure of the STAT3β homodimer co-crystallized with a DNA fragment. The two pockets interacting with the residues pTyr705 and Leu706 of the phosphopeptide are highlighted.
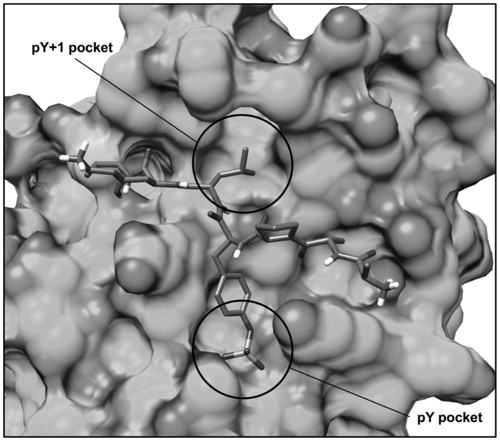
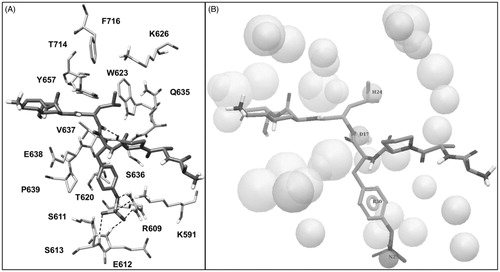
The phosphotyrosine pTyr705 of the peptide, that is essential for STAT3 dimerization, forms direct interactions through its phosphate group with Lys591, Arg609, Ser611, Glu612 and Ser613, constituting the so called pY pocket (); moreover, its aromatic ring well adapts into the narrow space delimited by Val637, Glu638 and Pro639 from one side and Lys591 from the other one. The peptide Leu706 forms an H-bond with the backbone oxygen of Ser636 through the amide N–H group and places its lateral chain into a hydrophobic pocket constituted by Trp623, Gln635, Val637, Tyr657, Thr714 and Phe716, also referred to as pY + 1 pocket (). Based on these considerations, we built a structure-based pharmacophore taking into account the interactions established by pTyr705 and Leu706 of the phosphopeptide. The model, shown in , was generated using the software PHASECitation22 and comprised (a) a negative feature representing the phosphate group of pTyr705, (b) an aromatic feature representing the aromatic ring of pTyr705, (c) an H-bond donor feature representing the amide N–H moiety of Leu706 interacting with Ser636 and (d) a hydrophobic feature representing the lateral chain of Leu706. In addition, the pharmacophore model was refined by adding excluded volume spheres mimicking the steric hindrance represented by the SH2 domain surface residues in proximity of the whole AAPpYLKT peptide (see “Materials and methods” section for details).
Figure 3. (A) Structure of the phosphopeptide AAPpYLKT (dark grey) bound to the STAT3 SH2 domain; the residues of the SH2 domain interacting with the pTyr705-Leu706 portion of the peptide and delimiting the pY pocket and the pY+1 pocket are shown (light grey). The main H-bonds detected in the complex are highlighted as black dashed lines. (B) Receptor-based pharmacophore model used for the VS study; the reference peptide (stick) together with the negative (N27), aromatic (R30), H-bond donor (D17) and hydrophobic (H24) features are shown; the excluded volume spheres are shown in light grey.
The pharmacophore model was used to screen the Vitas-M database, comprising about 1 320 000 commercially available compounds, retrieving only compounds matching all the four pharmacophoric features and respecting the volume constraints given by the SH2 binding site. By applying this strict filter, only 4819 compounds were further considered and subjected to the docking studies.
The extra precision method (XP) of GLIDE softwareCitation23 proved to be a particularly reliable docking procedure, according to our previous cross-docking studies carried out on different protein targetsCitation32,Citation33. For this reason, it was used as a first docking step to select the compounds that were more likely to bind the STAT3 SH2 domain by assuming a disposition matching the features of the pharmacophore model. The compounds were thus docked into the SH2 domain of the STAT3β monomer (PDB code 1BG1Citation20) and their top-scored poses were then superimposed to the pharmacophore model (see “Materials and methods” section for details) to select only the compounds matching all the four pharmacophoric features in their predicted binding mode. On these bases, 558 compounds were retained and subjected to further docking studies.
For this second analysis, we used a robust procedure employing AUTODOCK4Citation25 with customized docking parameters (see “Materials and methods” section for details), which was calibrated by docking the pTyr705-Leu706 dipeptide into the STAT3 SH2 domain. As shown in , the procedure was able to reproduce the binding disposition adopted by pTyr705 and Leu706 residues within the phosphopeptide AAPpYLKT with high accuracy, thus allowing a perfect matching of the four pharmacophoric features. The 558 ligands selected through the docking with GLIDE were thus further docked using this thorough AUTODOCK procedure and then filtered based on the superimposition to the pharmacophore model as in the previous docking step. The 65 compounds still matching the four pharmacophoric features of the model were selected and subjected to MD simulations in order to verify the stability of the their docking poses.
Figure 4. Structure of the pTyr705-Leu706 dipeptide (black stick) docked into the STAT3 SH2 domain (shown as protein surface) superimposed to the bound phosphopeptide AAPpYLKT (dark grey stick).
Before setting up the simulation protocol, the missing portion of the loop connected to the STAT3 SH2 domain, corresponding to residues P689-S701, was reconstructed using Modeller softwareCitation27; then, only the sequence corresponding to residues M586-F716, comprising the SH2 domain and the reconstructed loop, was taken into account for the MD simulation study. The simulation protocol was set up using the complex of the phosphopeptide AAPpYLKT bound to the SH2 domain of STAT3 as a test. The complex was subjected to a total of 2 ns of MD simulation and the total energy of the system during the simulation was analyzed; after about 400 ps, the system attained an equilibrium, since during the following 1.6 ns its total energy was found to be approximately constant (Supplemental S1). The analysis of the root-mean-square deviation (RMSD) of the receptor heavy atoms during the simulation, with respect to the starting structure, showed an initial increase due to the equilibration of the system followed by a stabilization of the RMSD value around 1.4 Å after about 400 ps. By the analysis of the RMSD of the disposition of the peptide during the simulation with respect to the input structure, we found an average RMSD of 2.0 Å. However, the portion of the peptide corresponding to our pharmacophore model and constituted by pTyr705 and Leu706 showed an average RMSD of 1.2 Å, highlighting that the positions of these two residues were well maintained during the whole simulation as well as their key interactions with the SH2 domain (Supplemental Figure S1). Precisely, the H-bond analysis revealed that the phosphate group of pTyr705 formed a total of 6 H-bonds with Arg609, Ser611, Glu612 and Ser613 that were maintained for almost the whole simulation. Similarly, the H-bond between the amide N–H group of Leu706 and the backbone oxygen of Ser636 was maintained for 97% of the simulation (Supplemental Table S1).
The 65 compounds selected through the docking step were subjected to the same MD simulation protocol described above and the average RMSD of their disposition during the simulation, with respect to their initial docking pose, was calculated. All the ligands showing an average RMSD > 2.0 Å were rejectedCitation34 and for the 19 remaining compounds the stability of the H-bonds formed with Ser636 and with the residues defining the pY pocket were analyzed. The four compounds maintaining an H-bond with the backbone oxygen of Ser636 and an H-bond with at least two pY pocket residues among Arg609, Ser611, Glu612 and Ser613 for more than 80% of the whole MD simulation were selected to be purchased and subjected to STAT3 SH2 binding assays. shows the biological results for the four purchased compounds, together with the reference compound MD77Citation29.
Table 1. Structure and STAT3 inhibitory activity of the tested compounds.
Compound VS1, which showed the highest inhibitory activity toward STAT3, was subjected to further assays for the determination of the IC50 for STAT3 as well as for STAT1 and Grb2, which present a high percentage of sequence homology with STAT3 (78 and 65%, respectively). As shown in , compound VS1 showed an IC50 value for STAT3 of 169.2 μM; moreover, the compound proved to be significantly selective versus Grb2, showing a complete lack of Grb2 inhibitory activity at the concentration of 200 μM (Supplemental Table S2).
Table 2. Structure and IC50 values toward STAT3, STAT1 and Grb2 of compounds VS1 and MD77.
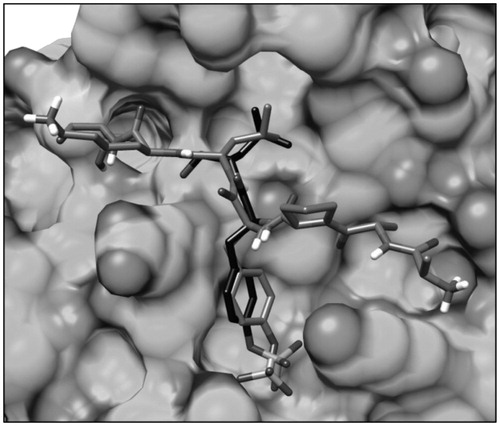
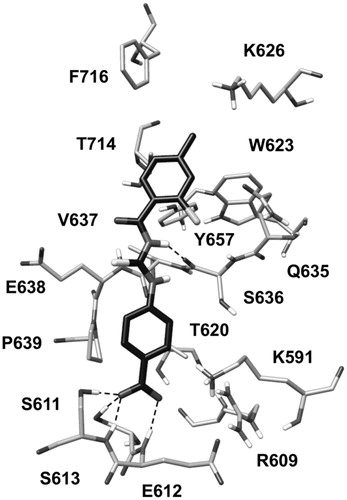
As shown in , compound VS1 well mimics the portion of the phosphopeptide constituted by pTyr705 and Leu706. The carboxylic group of the ligand is placed within the pY pocket and it predominantly interacts with Ser611, Glu612 and Ser613, establishing four different H-bonds with these residues. Precisely, the compound forms an H-bond with the hydroxyl group of Ser611, one with the backbone N-H of Glu612 and two H-bonds with Ser613 (one with the backbone and one with the hydroxyl group). The phenyl ring of the ligand linked to the carboxylic group takes contact with Thr620, Glu638 and Pro639, while the central moiety interacts with Val637 and forms an H-bond with Ser636 that is maintained for more than 95% of the whole MD simulation. Finally, the 2-chloro-4-methylphenyl portion of the ligand is well placed into the hydrophobic pocket constituted by Trp623, Gln635, Val637, Tyr 657, Thr714 and Phe716, forming Van der Waals interactions with these residues as well as with the side chains of Lys626 and Gln635.
Figure 5. Minimized average structure of compounds VS1 docked into STAT3 SH2 domain.
Conclusions
In this study, we developed a VS study aimed at the identification of new inhibitors of STAT3 dimerization. For this purpose, we built a receptor-based pharmacophore model comprising the key interactions established between the SH2 domains of the two STAT3 monomers within the STAT3 homodimer, which was used to screen a commercial database of about 1 320 000 compounds. The filtered ligands were then subjected to a docking analysis, employing two different docking procedures, as well as to MD simulation studies, allowing the selection of 4 candidates that were purchased and subjected to STAT3 SH2 binding assays. One of the selected compounds showed a significant inhibitory activity toward STAT3 and selectivity versus Grb2, thus validating the reliability of the VS approach herein described. Moreover, such compound could already be considered as a lead for the development of new and more potent STAT3 dimerization inhibitors targeting additional receptor subpockets that have not been taken into account in this study.
Supplementary material available online
Supplementary Figure S1 and Tables S1-S2
IENZ_1079184_Supp.pdf
Download PDF (276.7 KB)Acknowledgements
Many thanks are due to Prof. Maurizio Botta for using PHASE and GLIDE program in his computational laboratory (University of Siena, Italy) and warmly thank Prof.ssa Daniela Barlocco for the helpful discussion.
Declaration of interest
The authors report no declarations of interest. The authors acknowledge the Italian Ministero dell’Università e della Ricerca (MIUR), under the National Interest Research Projects framework (PRIN_2010_5YY2HL) for the financial support.
Related Research Data
References
- Darnell JE Jr. Transcription factors as targets for cancer therapy. Nat Rev Cancer 2002;2:740−9
- Darnell JE Jr. STATs and gene regulation. Science 1997;277:1630−5
- Turkson J, Jove R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 2000;19:6613−26
- Bromberg J, Darnell JE Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000;19:2468−73
- Turkson J. STAT proteins as novel targets for cancer drug discovery. Expert Opin Ther Targets 2004;8:409–22
- Masciocchi D, Gelain A, Villa S, et al. Signal Transducer and activator of transcription 3 (STAT3): a promising target for anticancer therapy. Future Med Chem 2011;3:367–97
- Debnath B, Xu S, Neamati, N. Small molecule inhibitors of signal transducer and activator of transcription 3 (STAT3) protein. J Med Chem 2012;55:6645−68
- Costantino L, Barlocco D. STAT3 as a target for cancer drug discovery. Curr Med Chem 2008;15:834–43
- Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci USA 2005;102:4700−5
- Siddiquee K, Zhang S, Guida WC, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA 2007;104:7391−6
- Matsuno K, Masuda Y, Uehara Y, et al. Identification of a new series of Stat3 inhibitors by virtual screening. ACS Med Chem Lett 2010;1:371−5
- Hao W, Hu Y, Niu C, et al. Discovery of the catechol structural moiety as a Stat3 SH2 domain inhibitor by virtual screening. Bioorg Med Chem Lett 2008;18:4988–92
- Xu X, Kasembeli MM, Jiang X, et al. Chemical probes that competitively and selectively inhibit Stat3 activation. PLoS One 2009;4:e4783–94
- Zhang M, Zhu W, Li Y. Discovery of novel inhibitors of signal transducer and activator of transcription 3 (STAT3) signaling pathway by virtual screening. Eur J Med Chem 2013;62:301–10
- Liu LJ, Leung KH, Chan DS, et al. Identification of a natural product-like STAT3 dimerization inhibitor by structure-based virtual screening. Cell Death Dis 2014;5:e1293–302
- Chen H, Yang Z, Ding C, et al. Fragment-based drug design and identification of HJC0123, a novel orally bioavailable STAT3 inhibitor for cancer therapy. Eur J Med Chem 2013;62:498–507
- Yu W, Xiao H, Lin J, Li C. Discovery of Novel STAT3 Small molecule inhibitors via in silico site-directed fragment-based drug design. J Med Chem 2013;56:4402−12
- Shahani, VM, Yue P, Haftchenary S, et al. Identification of purine-scaffold small-molecule inhibitors of stat3 activation by QSAR studies. ACS Med Chem Lett 2011;2:79–84
- Leung KH, Liu LJ, Lin S, et al. Discovery of a small-molecule inhibitor of STAT3 by ligand-based pharmacophore screening. Methods 2015;71:38–43
- Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature 1998;394:145−51
- Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res 2000;28:235–42
- PHASE, version 3.1. New York (NY): Schrödinger LLC; 2009
- GLIDE, version 5.0. Portland (OR): Schrödinger Inc; 2009
- Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model 1999;17:57–61
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91
- Case DA, Darden TA, Cheatham III TE, et al. AMBER, version 11. San Francisco (CA): University of California; 2010
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993;234:779–815
- York DM, Darden TA, Pedersen LG. The effect of long-range electrostatic interactions in simulations of macromolecular crystals – a comparison of the Ewald and truncated list methods. J Chem Phys 1993;99:8345–8
- Masciocchi D, Villa S, Meneghetti F, et al. Biological and computational evaluation of an oxadiazole derivative (MD77) as a new lead for direct STAT3 inhibitors. Med Chem Commun 2012;3:592–9
- Uehara Y, Mochizuki M, Matsuno K, et al. Novel high-throughput screening system for identifying STAT3-SH2 antagonist. Biochem Biophys Res Commun 2009;380:627–31
- Turkson J, Ryan D, Kim JS, et al. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem 2001;276:45443–55
- Tuccinardi T, Poli G, Romboli V, et al. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J Chem Inf Model 2014;54:2980−6
- Tuccinardi T, Poli G, Dell'Agnello M, et al. Receptor-based virtual screening evaluation for the identification of estrogen receptor β ligands. J Enzyme Inhib Med Chem 2015;30:662−70
- Poli G, Giuntini N, Martinelli A, Tuccinardi T. Application of a FLAP-consensus docking mixed strategy for the identification of new fatty acid amide hydrolase inhibitors. J Chem Inf Model 2015;55:667–75