
Abstract
Therapeutic monoclonal antibodies targeting G-protein-coupled receptors (GPCRs) are desirable for intervention in a wide range of disease processes. The discovery of such antibodies is challenging due to a lack of stability of many GPCRs as purified proteins. We describe here the generation of Fpro0165, a human anti-formyl peptide receptor 1 (FPR1) antibody generated by variable domain engineering of an antibody derived by immunization of transgenic mice expressing human variable region genes. Antibody isolation and subsequent engineering of affinity, potency and species cross-reactivity using phage display were achieved using FPR1 expressed on HEK cells for immunization and selection, along with calcium release cellular assays for antibody screening. Fpro0165 shows full neutralization of formyl peptide-mediated activation of primary human neutrophils. A crystal structure of the Fpro0165 Fab shows a long, protruding VH CDR3 of 24 amino acids and in silico docking with a homology model of FPR1 suggests that this long VH CDR3 is critical to the predicted binding mode of the antibody. Antibody mutation studies identify the apex of the long VH CDR3 as key to mediating the species cross-reactivity profile of the antibody. This study illustrates an approach for antibody discovery and affinity engineering to typically intractable membrane proteins such as GPCRs.
Abbreviations
| GPCR | = | G-protein coupled receptor |
| scFv | = | single chain Fv fragment |
| IgG | = | immunoglobulin G |
| CDR | = | complementarity determining region |
| FPR | = | formyl peptide receptor |
| FMAT | = | fluorometric microvolume assay technology |
| FLIPR | = | Fluorescent Imaging Plate Reader |
| Fv | = | variable domain |
| MPL | = | magnetic proteoliposome |
| RIMMS | = | repetitive immunization at multiple sites |
| VH | = | variable heavy |
| VL | = | variable light |
Introduction
The formyl peptide receptor (FPR) family is a group of Class A G-protein coupled receptors (GPCRs) that mediate leukocyte responses during inflammation.Citation1-Citation4 In humans, the FPR receptor family is composed of 3 members: FPR1, FPR2/ALX and FPR3. Human FPR1 is primarily expressed in neutrophils, monocytes and macrophages and mediates effects such as degranulation and chemotaxis in response to a range of formyl-peptide ligands derived from bacteria and mitochondria. Therefore, FPR1 is a potential therapeutic target for the treatment of inflammation-related diseases that are exacerbated by bacterial infection and tissue damage.Citation5 In addition, FPR1 was shown to be selectively expressed on highly malignant human glioma cells and contribute to tumor progression, and therefore may be a potential therapeutic target for the treatment of malignant human glioblastoma.Citation6 A significant advantage offered by monoclonal antibodies over other classes of therapeutic for targeting GPCRs is their high specificity that allows precise selectivity for desired GPCR family members and even desired conformations of a particular GPCR. Therefore, monoclonal antibodies offer the possibility for identifying GPCR-targeting therapeutics with very specific functions and correspondingly few unwanted effects.Citation7 However, GPCRs and other complex integral membrane proteins are difficult targets for antibody isolation and affinity maturation. A limiting factor is typically the availability of a suitable protein preparation for use as an immunogen or antigen in antibody generation and optimization approaches. There are reports describing the use of purified GPCR preparations for antibody isolation, for example the immunization of animals with the rat neurotensin 1 receptorCitation8 and the rat 5HT2c serotonin receptor;Citation9 however, this is most often not the case and surrogate immunogens or antigens are usually required.Citation10 Peptides and proteins have been used successfully to mimic GPCR extracellular regions for antibody generation, but this approach is not applicable to all GPCRs because it depends on the size, the functional relevance and the sequence homology of these regions to other GPCRs in each case. Furthermore, the use of peptides is limited to mimicking linear epitopes rather than allowing the representation of conformational epitopes made up of multiple extracellular loops. Thyroid stimulating hormone receptor is an example of a class A GPCR very well suited to a peptide antigen approach since it possesses a large ectodomain that can be exploited for the design of immunogens, and, as a result, a panel of antibodies targeting this GPCR exist.Citation11 Other successful examples of the use of peptides as antigens include the isolation of single chain Fv fragment (scFv) antibodies that are able to recognize the native cholecystokinin-B receptor expressed on cells by panning of a phage display antibody library using a synthetic peptide corresponding to the second extracellular loop of the receptor,Citation12 and isolation of a competitive antagonistic antibody for the class B GPCR glucose-dependent insulinotropic polypeptide receptor using phage display and ribosome display selections against a peptide comprising the receptor N-terminus.Citation13 In cases where the use of extracellular domains or peptides as surrogate GPCR immunogens is not feasible, approaches that avoid the need for a purified GPCR have been used, such as the immunization of animals with DNA encoding the GPCR of interestCitation14 or the use of GPCR over-expressing cell lines for the immunization of animals. The latter is a widely used solution to identify antibodies targeting GPCRs, and by this route neutralizing antibodies to the CXCR4 receptor, the rat sphingosine 1-phosphate receptor and the CCR5 receptor among others have been derived.Citation15-Citation17 A recent publication describes the use of intact cells for the isolation of anti-chemokine receptor 4 (CCR4) antibodies from a naïve phage display human scFv library.Citation18
Therapeutic antibodies are often required to be of higher affinity than is typical for antibodies derived by immunization of animals or selected from naïve in vitro display libraries; however, in vitro affinity maturation of an antibody is ideally carried out using a purified source of the target protein that is structurally and functionally relevant to ensure antibodies with the desired activity are obtained.Citation19,Citation20 Therefore, affinity optimization of antibodies targeting complex integral membrane proteins such as GPCRs, particularly those with relatively small extracellular regions, is a significant challenge limiting the availability of suitable antibodies to modulate this important group of potential therapeutic targets. FPR1 has small extracellular loops and is unstable in purified form; therefore, it can be seen to exemplify a typically challenging GPCR for antibody generation and engineering. Here we describe an anti-FPR1 human monoclonal antibody derived by immunization of transgenic mice with whole cells, followed by engineering for increased potency and species cross-reactivity using phage display and whole cells as the sole source of FPR1 antigen. This approach may be applicable for the discovery of antibodies targeting other complex integral membrane drug targets.
Results
Identification of the anti-FPR1 antibody Hy38-1
Hy38-1 is a human monoclonal antibody specific for human FPR1 that was derived by immunization of transgenic mice expressing human variable region genes. Species cross-reactivity of potential therapeutic antibodies, in particular functional inhibition of the antibody target in cynomolgus monkey, is typically desirable to facilitate preclinical safety studies.Citation21 For FPR1, this is a challenge because there is relatively little of the protein exposed at the cell surface, and the 75% amino acid sequence identity of these small extracellular regions between human and cynomolgus monkey is relatively low. Therefore, an immunization and screening strategy to increase the likelihood of identifying an antibody with species cross-reactivity to cynomolgus FPR1 was employed to generate an anti-FPR1 antibody for development as a potential therapeutic. Mice were immunized with whole cells over-expressing human FPR1 in 2 different cell backgrounds (CHO-K1 and HEK293) or alternately with cells over-expressing human FPR1 and cynomolgus FPR1. Hybridoma supernatants were assayed for specific binding to human and cynomolgus FPR1-over-expressing cells. Variable region genes from hybridomas were cloned into expression vectors for the production of full-length purified human IgG1, and antibodies were then tested in functional cell-based assays for the ability to inhibit formyl peptide-induced responses.
From a total of 36,960 hybridoma wells screened (of which approximately 30% were confirmed as containing IgG-expressing hybridomas; data not shown), 192 showed specific binding to either human or cynomolgus FPR1 expressing cells, but not parental cells, and a further 13 were identified as binders to both human and cynomolgus FPR1 expressing cells. DNA sequencing of the variable regions showed the majority of these antibodies were different (>95% diversity based on VH sequences, >80% diversity based on VL sequences). From the 13 cross-reactive binders, 12 were unique and only a single antibody (Hy38-1) showed functional inhibition of both human and cynomolgus FPR1, albeit with relatively low potency and in particular only very weak activity for cynomolgus FPR1. Hy38-1 was able to inhibit fMIFL-induced calcium signaling in human FPR1 Gα16-coupled transfected HEK cells with an IC50 of 36.5 nM against 300 pM fMIFL. The most potent human FPR1-specific antibody identified had an IC50 of 87 nM against 10 nM fMIFL, while Hy38-1 showed no inhibition at this higher ligand concentration. Hy38-1 did not show any activity in the equivalent cynomolgus FPR1 calcium release assay. Notably, Hy38-1 did show a trace of activity in a primary neutrophil CD11b up-regulation assay at the highest concentration of antibody tested (83% of the maximum 1 nM fMLFF signal at 270 nM Hy38-1, compared to an IC50 of 34 nM in the equivalent human assay). The most potent human FPR1 active antibody was derived from the modified RIMMS (repetitive immunization at multiple sites) immunizations using human FPR1 only; Hy38-1 also came from this approach. The weak inhibition of formyl peptide-induced cynomolgus neutrophil activation seen in response to Hy38-1 suggested the potential for improved potency upon affinity maturation. Therefore, affinity maturation of Hy38-1 was carried out to improve its potency for both human and cynomolgus FPR1.
Potency optimization of Hy38-1
CDR-targeted mutagenesis
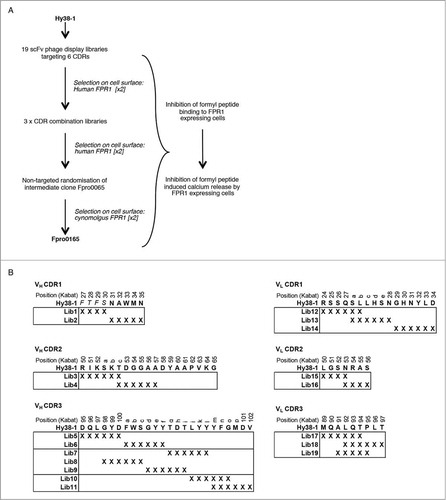
An overview of the selection and screening cascade to improve the potency of Hy38-1 against both human and cynomolgus FPR1 is illustrated in . The CDRs of Hy38-1 (defined by Kabat et al.Citation22 and numbered accordingly throughout) are shown in . At 24 residues, the VH CDR3 of Hy38-1 is longer than average for a human antibody, in which the VH CDR3 regions are more typically in the region of 8 to 18 amino acids.Citation23 Longer VH CDR3 regions have been suggested to be important for binding to certain target classes, such as GPCRs, where antigen binding might be facilitated by a longer projecting structure.Citation24 Interestingly, the aforementioned human FPR1-specific antibody isolated by immunization also had long VH CDR3 region (22 amino acids). We therefore considered that the VH CDR3 of Hy38-1 could be particularly critical for FPR1 binding, and so, in the design of the targeted mutagenesis approach, we allowed the VH CDR3 to be very thoroughly explored in a range of libraries. In addition, we extensively targeted the other 5 CDRs in the event that the VH CDR3 region was not amenable to change. Nineteen individual Hy38-1 scFv phage display libraries targeting all 6 CDRs were constructed (). In each library, 4 to 6 consecutive codons were replaced by NNS codons to allow for the inclusion of any amino acid at these targeted positions. The VH CDR3 and VL CDR3 sequences were covered by several overlapping libraries to allow for the potential for mutations of the majority of residues in more than one context, for example VH CDR3 position 98 was varied in the context of changes spanning positions 95 to 100, as well as in the context of changes spanning residues 98-100c. Prior to selection, libraries were pooled by CDR to a maximum of 3 mutagenesis blocks per pool, and then these pooled libraries were selected for 2 rounds on human FPR1 over-expressing cells.
Figure 1. Antibody engineering strategy (A) and selection and screening cascade (B) used for the optimization of Hy38-1. Amino acid residues of Hy38-1 are shown in uppercase with the 4 residues C-terminal to the VH CDR1 region shown in italics. X denotes replacement of a codon with the degenerate codon NNS. The individual libraries within each CDR were pooled for selection as shown by the boxes. CDR residues are numbered according to Kabat et al.Citation22
After a second round of selection and screening of the CDR-targeted libraries, 12 scFv antibodies were chosen and reformatted as full-length IgGs. Three IgGs were identified as having an improved ability to inhibit the binding of fMLFK to human FPR1 and cynomolgus FPR1-expressing cells compared to Hy38-1 (). Two of these corresponded to Hy38-1 variants that were able to inhibit the fMIFL-induced calcium signaling in human FPR1 Gα16-coupled transfected HEK cells in scFv form (Fpro0024 scFv: 356 nM IC50, Fpro0025 scFv: 38 nM IC50). As full-length human IgG, all of the antibodies tested inhibited fMIFL-induced calcium signaling by human FPR1; however, none of the IgGs tested showed inhibition in the cynomolgus FPR1 calcium release assay (). The most improved antibody was Fpro0025, a variant of Hy38-1 containing mutations in VL CDR1 that was identified from the second round of phage display selection on cells. Fpro0025 had an approximately 40-fold lower IC50 than Hy38-1 in the human FPR1 calcium release assay.
Table 1. Amino acid sequence and potency of affinity matured FPR1-specific antibodies. Antibodies shown correspond to those from the second round of selection that were improved compared to Hy38-1 in the fMLFK binding assay, those from the fourth round of selection that showed inhibition in the cynomolgus calcium release assay, and Fpro0165, the cross-reactivity optimized variant identified after 6 rounds of selection
CDR combination and random mutagenesis
For further affinity optimization, 3 new libraries in which 2 CDRs were simultaneously randomized were prepared by combining the previously selected CDR mutant populations. This process produces new variants in which potentially additive or synergistic improvements can be achieved by the random pairing of selected CDR variants followed by further selection. Only VL CDR libraries were chosen for combination since these selected populations contained greater numbers of more potent inhibitors compared to the VH CDR libraries (). The 3 new VL CDR combination libraries, combining VL CDR1with VL CDR2, VL CDR2 with VL CDR3 and VL CDR1 with VL CDR3, were subjected to an additional 2 rounds of phage display selection on human FPR1-expressing cells.
Figure 2. Inhibition of fMLFK binding to human FPR1-expressing cells by crude scFv following 2 rounds of selection on human FPR1-expressing cells (A). The VH CDR3 randomized populations are absent since they were not progressed beyond the first round of selection due to a high abundance of stop codons. Each data point represents the single-point testing of an individual scFv for the ability to inhibit the binding of AlexaFluor-647-labeled fMLFK to FPR1-expressing cells. scFv are not purified or normalized for concentration. Data is expressed as a percentage of fMLFK binding in the absence of antibody and the mean percentage inhibition for each population is illustrated as a solid horizontal line. Complete inhibition of 400 pM fMLFF-induced calcium signaling in both human (B) and cynomolgus (C) FPR1-transfected HEK Ca2+ reporter cell assay by Fpro0165 purified scFv. Fpro165 was derived by random mutagenesis of Fpro0065 followed by phage display selection on cynomolgus FPR1 expressing cells, and was the only scFv identified with full activity in the cynomolgus FPR1 Ca2+ reporter cell assay. Fpro0025 scFv, which is inactive in the cynomolgus FPR1 calcium release assay but is of equivalent potency in the human FPR1 assay is shown for comparison.
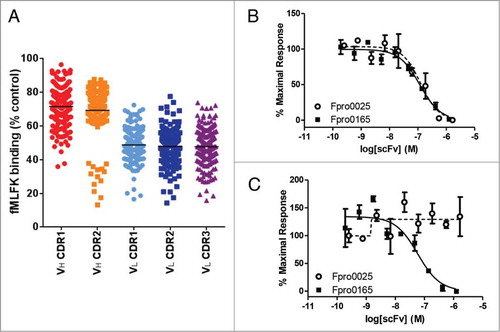
After periplasmic scFv screening of the VL CDR combination libraries, 55 scFvs showing greater inhibition of formyl peptide binding to FPR1-expressing cells than the parent Hy38-1 were identified and purified to determine IC50 values, and 25 of the most potent scFv were reformatted as full-length human IgG. Formyl peptide neutralizing activity in the cynomolgus FPR1 calcium release assay was seen for the first time for 3 of the 25 IgGs that were tested (). However, the IC50 values for inhibition of formyl peptide-induced calcium release on human FPR1 were at best only 2-fold improved compared to that of Fpro0025, despite CDR combination and further selection. Fpro0067 was the most improved IgG in the human FPR1 calcium release assay that importantly showed activity in the corresponding cynomolgus FPR1 calcium release assay. Interestingly, Fpro0067 is in fact identical to Fpro0025, the most potent VL CDR1 variant of Hy38-1, but now with additional amino acid changes in the VL CDR2 ().
To further increase affinity for cynomolgus FPR1, additional sequence diversity was introduced into Fpro0067 by random mutagenesis, and this library was then used for phage display selection on cynomolgus FPR1-expressing cells. This led to the isolation of antibody Fpro0165, which was the only variant of Hy38-1 identified throughout the optimization process to show inhibition in the cynomolgus FPR1 calcium release assay as a scFv (). Upon reformatting to an IgG, Fpro0165 showed a greatly improved potency in the cynomolgus FPR1 calcium release assay (). Fpro0165 has 2 mutations from Fpro0067 in the VH region: Ile to Val at Kabat position 51 (Ile51Val) in the VH CDR2, and Ser100cAsn in the VH CDR3.
Potency, cross-reactivity and specificity of Frpo0165
The potency of Fpro0165 IgG was assessed in further human cell-based assays to confirm that the potency improvements seen using human FPR1 and cynomolgus FPR1transfected cell lines translated to more potent inhibition of FPR1-mediated neutrophil effects compared to Hy38-1. Inhibition of primary human neutrophil chemotaxis in response to prototypical bacterial (fMLFF, fMILF) and mitochondrial (fMMYALF) formyl peptide ligands showed that the potency of Fpro0165 was improved 15 to 23-fold compared to Hy38-1. Hy38-1 inhibited fMLFF, fMILF and fMMYALF induced neutrophil chemotaxis with mean KB values of 7.3 nM, 25.6 nM and 6.0 nM, respectively, while the corresponding values for Fpro0165 were 0.47 nM, 1.13 nM and 0.31 nM, respectively. The degree of human and cynomolgus FPR1 cross-reactivity of Fpro0165 was determined by a pA2 analysis using an assay measuring CD11b upregulation in primary human and cynomolgus neutrophils as a measure of neutrophil activation in response to formyl peptide. This pharmacological analysis was used to estimate antibody affinity due to the lack of suitable purified FPR1 proteins for direct affinity measurements. We quote ‘apparent pA2’ to acknowledge that all the rules of the Schild model were not satisfied, for example the non-parallelism of the agonist dose-response curves at different concentrations of antagonist (). The shape and steepness of the curves, along with their non-parallelism, might support a non-competitive inhibition of the agonist binding pocket by Fpro0165; however, this remains speculative without further mathematical and mechanistic modeling of the interaction. These apparent K d measurements can be used to support the conclusion that Fpro0165 has similar affinities for both human FPR1 and cynomolgus FPR1 (6.2 nM and 4.1 nM, respectively; ).
Figure 3. Determination of the apparent affinity of Fpro0165 for human FPR1 and cynomolgus FPR1 using Schild analysis of activity in a CD11b upregulation assay using in primary human and cynomolgus neutrophils. Concentration-response curves of fMLFF induced CD11b expression in primary human (A) and cynomolgus (B) neutrophils in the absence (0) or presence of different concentrations Fpro0165 IgG (indicated in the key as logM). Schild plot analysis of the data derived from human (C) and cynomolgus (D) assays to generate the apparent Kd of the antibody. Dose ratios were generated using 50% response levels of fMLFF in the presence of antibody and log (dose ratio-1) was plotted against log [antibody] (M). The x-intercept of the fitted regression line is an estimate of pA2, the apparent equilibrium dissociation constant for the antibody.
![Figure 3. Determination of the apparent affinity of Fpro0165 for human FPR1 and cynomolgus FPR1 using Schild analysis of activity in a CD11b upregulation assay using in primary human and cynomolgus neutrophils. Concentration-response curves of fMLFF induced CD11b expression in primary human (A) and cynomolgus (B) neutrophils in the absence (0) or presence of different concentrations Fpro0165 IgG (indicated in the key as logM). Schild plot analysis of the data derived from human (C) and cynomolgus (D) assays to generate the apparent Kd of the antibody. Dose ratios were generated using 50% response levels of fMLFF in the presence of antibody and log (dose ratio-1) was plotted against log [antibody] (M). The x-intercept of the fitted regression line is an estimate of pA2, the apparent equilibrium dissociation constant for the antibody.](/cms/asset/cc621d59-82df-4171-99fa-ebf94251cc23/kmab_a_985158_f0003_c.jpg)
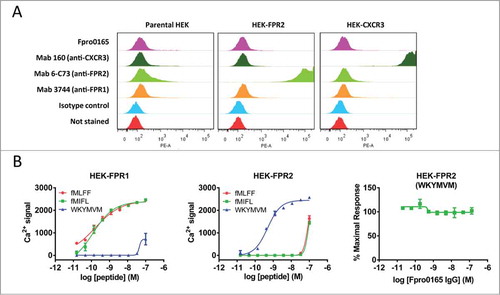
FPR1 is one of 3 members of the FPR family, alongside FPR2, which has 69% overall sequence identity to FPR1 (52% identity in the extracellular loops), and FPR3, which has 59% overall sequence identity to FPR1 (40% identity in the extracellular loops). Specificity of Fpro0165 for FPR1 was confirmed by lack of binding to cells over-expressing the most closely related family member FPR2 using FACS, and by lack of activity in a human FPR2 calcium release assay. Minimal staining on HEK cells expressing FPR2 was demonstrated by Fpro0165, while strong staining was obtained on those same cells using an FPR2-specific antibody (). Very weak staining by Fpro0165 seen in FPR2-expressing cells was also seen to a similar level on the HEK parental cells or HEK expressing another GPCR (human CXCR3). Therefore, we suggest this staining is not due to FPR2 binding, but most likely corresponds to low basal expression of FPR1 on the HEK cell line background. This is consistent with the same low level of staining obtained when using the commercially available FPR1 antibody. The FPR2-specific formyl peptide WKYMVM was able to induce calcium release in the human FPR2 expressing cell line; however, this was not inhibited by Fpro0165, confirming specificity of Fpro0165 for FPR1 over FPR2 ().
Figure 4. Specificity of Fpro0165 for FPR1. FACS staining was carried out using Fpro0165 IgG1 and control IgG1 with parental, human FPR2-overexpressing and human CXCR3-overexpressing HEK cells to determine the binding specificity of Fpor0165 to FPR1 (A). See Results text for details. Lack of inhibition of WKYMVM-induced calcium release in FPR2-overexpressing cells by Fpro0165 (B, right panel). Different agonist activity of the FPR1-specific formyl peptides fMLFF (EC50 not quoted, incomplete curve) and fMIFL (EC50, 0.12 nM on FPR1 cells) and FPR2-specific peptide WKYMVM (EC50, 0.49 nM on FPR2 cells) are shown in the left and center panels confirming the overexpression of FPR1 and FPR2 respectively.
Crystal structure of Fpro0165
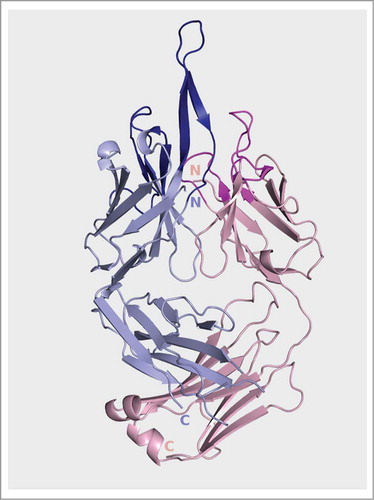
The crystal structure of Fpro0165 Fab was solved in order to determine the conformation of the long VH CDR3. The data was anisotropic with diffraction to 3.1 Å in the best direction, but only 3.7 in the orthogonal direction. Electron density was clear for the main-chain of the entire VH CDR3, but only partial density was observed for the side-chains of residues Gln96, Leu97, Tyr99, Phe100a, Tyr100b, Tyr100e, Tyr100f, Leu100j, Tyr100k, Tyr100l, Tyr100m and Phe100n. The crystallographic data and refinement statistics are shown in . The structure shows that VH CDR3 forms an extended β-ribbon conformation protruding beyond the rest of the antigen combining surface formed by the other CDRs (). Interestingly, a similar secondary structure has previously been reported for an anti-HIV antibody having a long VH CDR3.Citation25
Table 2. Crystallography data collection and refinement statistics
Figure 5. Ribbon representation of the Fpro0165 Fv structure. Blue is heavy chain; pink is light chain.
Molecular modeling of the Fpro0165 Fab—FPR1 complex
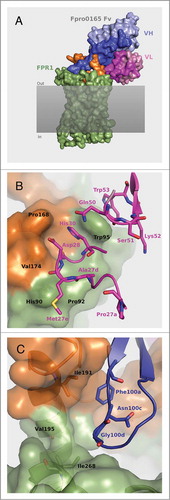
To investigate the hypothesis that the long VH CDR3 loop of Fpro0165 mediates interactions with FPR1, and to potentially rationalize the amino acid changes seen upon optimization of Hy38-1, the crystal structure of Fpro0165 Fab was used for computational protein-protein docking with a homology model of human FPR1 (). In the model of the Fpro0165 Fv – FPR1 complex, the VH CDR3 buries substantial surface area across the extracellular loop 2 of the receptor. VL CDR1 and VL CDR2 residues contribute to the remainder of the buried surface area at the interface with the receptor. The Fpro0165 VL CDR2-optimized residues Gln50, Ser51, Lys52 and Trp53 surround FPR1 residues Trp95, along with the VL CDR1 residue His30 that was unchanged during optimization (). VL CDR2 Trp53 is predicted to form pi stacking interactions with FPR1 Trp95. The optimized VL CDR1 residues His27dAla, Ser27eMet and Asn28Asp are predicted to optimize Van der Waals interactions with FPR1. The VL CDR1 Ser27aPro mutation is not predicted to be part of the paratope region. The complex also suggests that the VH CDR2 mutation Ile51Val does not form part of the binding region, while, in contrast, the VH CDR3 mutation Ser100cAsn is located proximal (within 6Å) to a proposed interaction within a hydrophobic region formed by Ile191 and Val195 of human FPR1 and Phe100a of VH CDR3 (). In cynomolgus FPR1, these 2 residues are substituted by Leu193 and Ile197, respectively, which could modify the volume available for side chain packing at position 100 c of VH CDR3. While offering potential insight into the structural basis of inhibition, it is important to note that these observations should be regarded as putative, due to the speculative nature of homology modeling.
Figure 6. Structural model of the Fpro0165 Fv – FPR1 docked complex (A) and close up views of the optimized VL CDR amino acid residues (B) and the VH CDR3 Asn100c and Gly100d and surrounding region (C). For clarity in panels B and C, only the relevant residues are shown as sticks and labeled. Pale blue shows the heavy chain with CDR regions shown in darker blue; pink shows the light chain with CDR regions shown in magenta; green is FPR1; orange is the extracellular loop 2 region of FPR1; gray shaded area represents the cell membrane.
Functional analysis of VH CDR3 Asn100c and Gly100d in Fpro0165
Structural analysis of the predicted Fpro0165 Fv – FPR1 complex suggests that the apex of the VH CDR3 loop may be the major component of the antibody paratope and provides a hypothesis for the importance of the Ser100cAsn mutation and position Gly100d in mediating improved cynomolgus FPR1 neutralization of the Fpro0165 antibody. This was investigated experimentally by individual mutation of Asn100c and Gly100d to all other amino acids (with the exception of cysteine) to determine the effect on human FPR1 and cynomolgus FPR1 potency.
Six amino acid substitutions of Asn at VH position 100 c resulted in an approximately 2-fold or more increase in potency for human FPR1 neutralization compared to Fpro0165, including reversion of this position to Ser (as is present in Hy38-1), as well as replacement with the polar or hydrophobic amino acids Thr, Phe, Trp, Met or Leu (). Substitution with other non-charged hydrophobic amino acids (Val, Ile, Gly, Ala or Tyr) broadly maintains human FPR1 potency of Fpro0165, with Ala being the least active. Notably, substitution with any charged amino acid (Asp, Arg, His, Glu, or Lys), or with Gln or Pro results in complete loss of activity. Also, while 11 out of the 19 Fpro0165 variants with amino acid replacements at Asn100c retained human FPR1 potency, the majority of Asn100c amino acid substitutions resulted in loss of activity in the cynomolgus FPR1 assay, and in fact only Thr, Ser and Leu substitutions retained any residual cynomolgus FPR1 activity. Substitution of Gly100d with other amino acids was less well tolerated, with only 3 of the 19 Gly100d Fpro0165 variants having human FPR1 activity similar to Fpro0165 and no amino acid substitution at Gly100d retaining any cynomolgus FPR1 activity. The docked model positions Gly100d slightly closer to Leu270 of cynomolgus FPR1 than to the structurally equivalent Ile268 of human FPR1 () (distance of 3.7 Å measured between the Cα atom of Gly100d and Cδ1 of Leu270 compared to 4.1 Å between Cα atom of Gly100d and Cδ1 of I268 in the respective rotamers of the side-chains seen in the model). Considering the dynamic nature of proteins, Leu270 may not fit optimally in the interacting region with residues other than Gly at position 100d.
Table 3. Influence of Fpro0165 Asn100c and Gly100d mutations on potency
Discussion
Affinity maturation of antibodies using in vitro display techniques is a highly efficient and successful route by which to generate potent antibodies for therapeutic use.Citation20 A critical factor in the success of antibody generation is the quality and biological relevance of the target antigen that is used. Ideally, this should be presented in its native conformation and be stable to ensure antibodies with the required biological function are identified. Typically, purified recombinant antigen preparations are used for affinity optimization of antibodies, and for certain subclasses of GPCRs with large extracellular domains this approach is feasible. For example, a GPCR-specific neutralizing antibody has recently been identified by in vitro selection of a naïve antibody library using reducing concentrations of the receptor N-terminus during rounds of selection to enrich the selected populations for higher affinity antibodies.Citation13 This approach should be equally applicable to the in vitro selection of antibody libraries for affinity optimization. In addition, the directed evolution of a single-domain camelid antibody fragment (nanobody) recognizing the β2-adrenoceptor (β2-AR) in its agonist-bound conformation has recently been described.Citation26 In this work, a library containing variants of a nanobody with the desired conformational selectivity was displayed on yeast and selected using purified, detergent-solubilised, N-terminal FLAG-tagged and C-terminal truncated biotinylated β2-AR employing a selection strategy to enrich for higher affinity nanobodies that still recognized the conformation of interest. The selected nanobody has facilitated the crystallisation of the β2-AR T4 lysozyme fusion in its natural agonist activated conformation.
Although successful examples of the use of purified extracellular regions and purified receptors have been described, this approach cannot be generalized because many GPCRs such as FPR1 have small extracellular regions and are not stable in purified form. FPR1 is such a GPCR for which the generation of functional antibodies is limited by the lack of a purified reagent suitable for affinity optimization. In this study, we described the isolation of a human FPR1 neutralizing antibody with cross-reactivity to cynomolgus FPR1. Only one neutralizing antibody recognizing both human and cynomolgus FPR1 was isolated, but this was less potent than other human FPR1-specific antibodies. This rarity is most likely due to precise paratope requirements for cross-reactivity that are only met by a small number of antibodies. We subsequently optimized the affinity of this species cross-reactive antibody by the selection of phage displayed CDR-targeted and random mutagenesis scFv libraries on intact cells over-expressing FPR1. Membrane receptor over-expressing cells have recently been used as an antigen source to affinity mature an anti-transferrin receptor single-chain antibody by yeast display,Citation27 confirming the use of intact cells as a successful route for antibody optimization. A recent publication also reports the use of cell-based phage display selection to optimize scFv targeting CCR4, although the details are not described.Citation18 Partially purified membrane protein preparations, for example in which membrane proteins are maintained in reconstituted particles in the context of a phospholipid bilayer, may be a suitable alternative over purified receptors or whole cells for antibody identification. An example of this approach has been used to identify a CCR5 neutralizing monoclonal antibody by selection of a human scFv phage display library.Citation28 Other strategies to create GPCR preparations for drug discovery are also emerging, for example stabilised receptor proteins (StaRs) containing point mutations that not only allow extraction of the GPCR from the cell membrane by solubilisation in detergent, but also conformationally lock the receptor in an active or inactive stateCitation10 have recently been used successfully for in vivo immunization to generate functional β1-adrenoceptor antibodies.Citation29
While capabilities for generating purified GPCRs are increasingly being developed, cells with suitable expression of the receptor of interest are required as a starting point. The direct use of the particular GPCR-expressing cells for antibody generation and engineering removes the need to further manipulate the receptor from the cell, which is likely to require specialist expertise, case-by case optimization and may not be achievable for all receptors. Therefore whole cells over-expressing the receptor of interest may represent the current most feasible and accessible source of a GPCR, or other complex integral membrane protein, in its native state for the purpose of in vitro affinity maturation of antibodies. It should be borne in mind that a potential disadvantage to the use of over-expressing cells can be receptor internalization. In addition, a feature of using whole cells as an antigen source for antibody discovery from naïve libraries or immunized animals is generally a high level of non-specific antibodies due to the fact that the cell surface presents many more irrelevant epitopes than the protein of interest, which is present at a relatively low concentration. In the present study, 1.8% of the IgG-expressing hybridomas yielded antibodies binding FPR1, the majority of which were unique. Similarly, in the recently published example of CCR4-expressing cell-based selection of a naïve phage display scFv library, 1.1% of the scFv antibodies screened were specific for CCR4, although only 4 out of these 132 antibodies were unique.Citation18 For the in vitro affinity maturation of antibodies, however, non-specific background due to the use of cells is likely to be less of an issue since the starting point is an antibody already recognizing the protein of interest.
In this study, the potency improvement of the hybridoma-derived antibody Hy38-1 for human FPR1 occurred as a result of changes predominantly in the light chain variable regions. The VH CDR3 appeared to be already optimal in that it did not tolerate mutagenesis and selection for improved human FPR1 potency. Structural analysis showed a protruding structure of the VH CDR3 of Fpro0165 and computational protein-protein docking suggests this VH CDR3 may be the primary site of interaction with extracellular loop 2 of human FPR1. Therefore, in vivo affinity maturation during immunization may indeed have already resulted in Hy38-1 having an optimal VH CDR3 region for human FPR1. The majority of the optimized residues of the VL CDRs are predicted to interact with FPR1, and in particular the Leu50Gln, Gly51Ser, Ser52Lys and Asn53Trp in the VL of Fpro0165 may have been selected to maximize Van der Waals interactions and buried surface area with FPR1 Trp95. The VL CDR1 Ser27aPro mutation was not predicted to interact with FPR1 in the complex; however, the selection of Pro here may have been driven by virtue of its restricted conformations, which could potentially result in a structural benefits such as increased stability of the VL CDR1.
A key aspect of the optimization of Hy38-1 was the improvement of cynomolgus FPR1 potency by random mutagenesis of the intermediate optimized antibody Fpro0067 and selection on cynomolgus FPR1 cells. This led to 2 further amino acid changes: Ile51Val in VH CDR2 and Ser100cAsn in VH CDR3. The former position does not interact with FPR1 in the model; therefore, it may be a bystander mutation or perhaps may act to stabilize the antibody structure, as the Val side chain points toward the upper core of the VH. The Ser100cAsn mutation, however, is located at the apex of the VH CDR3 loop where the model suggests that Asn may pack better than Ser with cynomolgus FPR1 due to stereochemical advantages. An experimental structure of the complex would be required to definitively confirm the structural rationale described in this work, although this is technically challenging due to the quality and quantity of purified FPR1 locked in a single conformation required. It should also be noted that the extracellular loops of FPR1 can be conformationally flexible, and this is not represented in the homology model or in the docked model of the complex. Hence, we recognize that the docked model represents only one potential interaction mode between the antibody and FPR1 and the observations described based on homology modeling and docking are speculative. Nevertheless, the data showing the effect on potency of amino acid substitutions in VH CDR3 at positions 100c and 100d indicates these residues must satisfy a specific combination of side-chain size, stereochemistry and hydrophobic character, thus providing some validation to the molecular modeling described. Further validation of the structural hypotheses could be obtained by performing site-directed mutagenesis of the receptor followed by assessment of the ability of Fpro0165 to inhibit the functional response to formyl peptide in cells expressing FPR1 mutants. For example, the hypothesis that pi stacking interactions occur between VL CDR2 Trp53 and FPR1 Trp95 could be tested by mutation of FPR1 Trp95 to Phe, Ala and Arg. This type of approach combining structural modeling and mutagenesis studies may offer useful information to support the design and engineering of antibodies against this important class of therapeutic target.
Materials and Methods
Materials
AlexaFluor647-labeled fMLFK and non-labeled fMIFL and fMMYALF were custom synthesized (Cambridge Research Biochemicals). fMLFF and WKYMVM peptides were obtained from Bachem (H-4294 and H-5798 respectively).
Cell lines
For immunizations, phage display selections and formyl peptide binding inhibition assays, human FPR1-expressing cell lines were obtained from MSM Protein Technologies Inc. and cynomolgus FPR1 expressing cell lines were prepared in-house. These cell lines were generated by transfection of CHO or HEK293 cells with either human FPR1 (haplotype 1Citation30) or cynomolgus FPR1 (common variant), to generate the cell lines CHO-hFPR-STR-TREX (T-RexTM-CHO, Invitrogen; R718-07), HEK-hFPR-C9 and CHO cFPR V3 clone 21 (CHO-K1). Human FPR1 expression levels were determined by quantitation of antibody binding sites using FACS staining with the human FPR1-specific antibody Mab3744-PE (R&D Systems; MAB3744) in conjunction with a calibrated curve prepared using Quantum beads (QuantumTM Simply Cellular anti-Mouse IgG, Bangs Laboratory, 815B). Cynomolgus FPR1 expression levels were estimated from the degree of FACS shift upon staining with AlexaFluor647-labeled fMLFFK. Transfected CHO cell lines expressed between 5 and 9 × 105 FPR1 molecules at the cell surface. For assay of FPR neutralizing activity, HEK293 cells were co-transfected with vectors containing either human FPR1, human FPR2 or cynomolgus FPR1, alongside the human G-protein subunit Gα16, to create the FPR1 calcium-signaling reporter cell lines HEK293-hFPRGα16-Clone 1, HEK293- hFPRL-1Gα16-c5 and HEK293-cFPRGα16-Clone20 respectively. Expression of FPR1 and FPR2 in the transfected HEK cell lines was confirmed by demonstration of calcium release in response to formyl peptides. The cell line HEK-TREX-hCXCR3 c19 for use as a GPCR-expressing negative control in FACS analysis was obtained from MSM Protein Technologies Inc.
Immunization and hybridoma generation
VelocImmune II mice (Regeneron, NY) aged between 6 and 8 weeks were used for immunization following 2 strategies: (a) human FPR1 over-expressing cells alone (alternating CHO-hFPR-STR-TREX and HEK-hFPR-C9) or (b) alternating human FPR1 or cynomolgus FPR1 over-expressing cells (CHO-hFPR-STR-TREX and CHO cFPR V3 clone 21). The immunization strategies were each carried out following both a modified 28-day RIMMS protocolCitation31 and a 48 day rest/boost protocol. For the RIMMS method, mice were primed subcutaneously on day 0 with 1 × 107 cells resuspended in Freund's complete adjuvant (Sigma, F5881). Subsequent boosts contained 1 × 107 cells resuspended in Freund's incomplete adjuvant (Sigma, F5506), administered subcutaneously on days 7, 14, and 20, with a final boost of both cell types given intraperionteally on day 23 (2 × 107 cells in PBS). Animals were sacrificed on day 28, and spleens and lymph nodes were harvested. For the rest/boost protocol, mice were primed with 2 subcutaneous injections of 100 ul, containing a total of 1 × 107 cells resuspended in Freund's complete adjuvant. For subsequent boosts on days 14, 28, and 42, 1 × 107 cells were resuspended in PBS and injected intraperitoneally. A final boost of both cell types was administered intraperionteally on day 45 (2 × 107 cells in PBS). Animals were sacrificed on day 49, and spleens and lymph nodes were harvested. Up to 1 × 108 lymphocytes per mouse, derived from the lymph nodes and spleen, were used for fusions with 2 × 107 with SP2/0 myeloma cells using polyethylene glycol.Citation32 Hybridomas were grown in selective media and supernatants were harvested after 13 days and assayed for binding to cells and for the ability to inhibit formyl-peptide binding to FPR1-expressing cells by FMAT®. VH and VL mRNA was isolated from hybridomas using Oligo (dT)25 beads (Novagen, 69593-3) and reverse-transcribed using SuperScript III Reverse Transcriptase (Invitrogen, 18080085). cDNA was tailed with poly(G) using dGTP and terminal transferase (NEB; M0315S) and VH and VL cDNA regions were amplified separately using oligo (dC)25 and either a specific constant heavy or light chain primer. Hybridoma supernatants were assayed for IgG/IgM expression using an FMAT® binding assay. Streptavidin beads were coated with biotinylated anti-mouse IgG (Sigma, B7022) or anti mouse IgM (Sigma, B9265) and the presence of IgG or IgM in the supernatant was subsequently detected with Alexa647-labeled anti-mouse IgG (Invitrogen, A21236) or Dylight649-labeled anti-mouse IgM (Jackson ImmunoResearch,115-495-075) and quantified against known standards. IgG positive supernatants were classified to be above 0.15 μg/ml IgG.
Generation and selection of targeted mutagenesis libraries
For targeted mutagenesis of Hy38-1, the VH and VL coding regions were used to prepare a scFv construct for optimization in phage display format. Large, CDR-targeted scFv phage libraries (greater than approximately 109 variant scFvs) were created by oligonucleotide-directed mutagenesis in which degenerate codons (NNS) were used to fully randomize regions of 6 consecutive amino acid residue using Kunkel mutagenesis.Citation33 Multiple overlapping libraries were created to randomize the CDR residues as described in the text and . In total, 19 individual scFv phage libraries were prepared and pooled for selection to a maximum of 3 libraries per pool resulting in 8 library pools, and these were then selected in solution on CHO K1 cells over-expressing human FPR1. Briefly, selections involved pre-blocking of phage and cells (approximately 5.0 × 106 cells per selection) for 1 h with 3% (v/w) non-fat skimmed milk powder in growth media, and then incubation of phage with cells for 2 h at room temperature. Six to 8 washes to remove non-bound phage were carried out by brief centrifugation to pellet the cells and gentle suspension of cells in PBS. Bound phage were eluted using high pH (100 mM triethanolamine and subsequently neutralized with 1 M Tris pH8.0) and used to infect logarithmically growing TG1 cells. The selected phage were rescued with helper phage for a further round of selection. The numbers of phage recovered from selections on FPR1-expressing cells compared to the numbers recovered from selection on non-transfected cells, along with the percentage of selected antibody sequences that were free from stop codons and were therefore expressing and displaying full-length antibody fragments, were used as an indication of the success of the selection of each library. The VH CDR3 libraries were not progressed beyond a first round of selection on cells due to the presence of a high percentage (>80%) of stop codon-containing antibody sequences in the selected population. After 2 rounds of selection, individual CDR libraries were combined to form single libraries in which clones contained randomly paired randomized CDR regions, and 2 further rounds of selection were carried out on human FPR1-expressing cells.
Generation and selection of the non-targeted random mutagenesis library
A DNA fragment encoding the scFv region of Frpo0067 and containing random mutations was created by error-prone PCR using the DiversifyTM PCR Random Mutagenesis Kit (Clontech, 630703). This was used as a mutagenic oligonucleotide for phage library construction by Kunkel mutagenesis.Citation33 For the first round of selection, phage were selected by panning on a cynomolgus FPR1 expressing CHO-K1 cell monolayer in a single T75 flask to recover and enrich for all cynomolgus FPR1 cell-binding phage following random mutagenesis. Rescued phage were then selected for a second round on cynomolgus FPR1 expressing CHO-K1 cells in solution as described for the selection of CDR targeted libraries.
Expression and purification of individual scFv
Individual scFv from the selections were expressed in E.coli and tested as non-purified periplasmic extracts for the ability to inhibit fMLFK binding to human FPR1 and cynomolgus FPR1 expressing cells. Inhibitory scFv with unique sequences were expressed in E.coli and purified by affinity chromatography for IC50 determination in the fMLFK binding inhibition assay and also in a functional signaling assay measuring inhibition of formyl peptide-induced signaling in calcium-coupled human FRP1 and cynomolgus FPR1 reporter cell lines.
IgG1 and Fab production
Antibodies were converted from scFv to IgG format by subcloning the VH and VL domains into human IgG heavy chain and light chain expression vectors that were co-transfected into HEK293/EBNA mammalian cells for expression. IgG proteins were purified from the culture medium using Protein A chromatography. An IgG1 mutant lacking effector function was used (IgG1_TMCitation34) to avoid possible complications involving effector function in cell-binding antibodies. Fpro0165 Fab was prepared from Fpro0165 IgG by papain digestion.
Inhibition of formyl-peptide binding to cells by FMAT®
FMAT® technology was used to measure the ability of antibodies to inhibit the binding of Alexa647-labeled FMLFK peptide to FPR1-expressing cells. Test antibodies were combined with an approximate EC75 concentration of Alexa647-labeled fMLFK and human FPR1 or cynomolgus FPR1 expressing CHO cells and incubated at room temperature for 2 h, after which fluorescence was measured using an FMAT 8200 cellular detection system (Applied Biosystems). For high-throughput screening of scFv populations, non-purified, bacterially expressed scFvs were assayed at a single concentration and the percentage inhibition of formyl peptide binding to cells in the absence of antibody was calculated. For assay of purified scFvs and IgGs, samples were assayed at multiple antibody concentrations in duplicate and non-linear regression analysis of concentration-response curves was used to determine of IC50 values. Appropriate scFv and IgG isotype controls were included in all assays.
Formyl-peptide induced calcium signaling assays
FPR1 reporter cell lines, comprising HEK293 (ECACC; 85120602) cells transfected with human FPR1 or cynomolgus FPR1 in combination with the human G-protein subunit Gα16, were used to identify antibodies that were able to inhibit the activation of FPR1 by formyl peptides. Potency (EC50) of formyl peptides required to induce calcium signaling was within approximately 20-fold of the concentrations required to induce physiological responses in neutrophils. In the Ca2+ reporter cell lines, stimulation of FPR1 with formyl peptide leads to calcium release that was measured using a calcium-sensitive fluorophore (FLUO-4 NW Calcium Assay kit (Molecular Probes)) in a plate-based fluorescence detection system (FLIPR- tetra, Molecular Devices). Antibodies were added to human FPR1 or cynomolgus FPR1 reporter cells in assay loading buffer which also contained the calcium-sensitive FLUO-4 dye and probenecid, and incubated for 30 minutes at 37°C 5% CO2, and then for a further 30 min at room temperature. Formyl peptides were then added and fluorescence was measured for a period of 3 min, and peak Ca2+ signal and percentage maximal response were derived from the data. For FPR1 assays, the formyl peptides fMIFL and fMLFF were used. fMIFL was used at an approximately EC50 concentration initially and then the concentration was increased to approaching its EC80 concentration as antibodies increased in potency during optimization. For greater discrimination of FPR1 antibody activity of the most optimized antibodies, the more potent peptide fMLFF was used (approximately EC50 concentrations). For human FPR2 assays, the FPR2-selective peptide WKYMVM was used (approximately EC50concentration). See Results for the actual agonist concentrations used in each case. Appropriate isotype control IgG were included in all assays; an example is shown in Figure S2.
Granulocyte chemotaxis assays
Primary granulocytes were isolated from human and cynomolgus peripheral blood by dextran sedimentation to remove erythrocytes, followed by discontinuous Percoll gradient centrifugation, according to standard granulocyte preparation methodology. Cells were incubated with the test antibodies at 37°C 5% CO2 for 30 min and chemotaxis in response to formyl peptides was measured using the ChemoTx® Disposable Chemotaxis System (Neuro Probe; 206-5). Formyl peptides were added to the lower chamber of an assay plate at approximately EC50 concentrations and cells pre-incubated with antibody were added to the top chamber. Plates were incubated at 37°C 5% CO2 for 90 min after which the cells that had migrated to the lower chamber were detected using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega; G7570). Appropriate isotype control IgG were included in all assays.
Primary neutrophil CD11b up-regulation assay
CD11b is a cell-surface protein that is up-regulated to neutrophil extracellular membrane upon activation, and was used as a marker to determine the ability of antibodies to inhibit the activation of primary neutrophils in response to formyl peptide. fMLFF was used for this assay as it was found to be the most potent of the formyl peptides tested. Primary human and cynomolgus granulocytes were incubated with antibodies at 37°C 5% CO2 for 30 min and were then washed and incubated with an approximately EC50 concentration of fMLFF at 37°C 5% CO2 for 1 h. Cells were then resuspended in 100 μL FACS buffer (PBS with 2% FBS) containing 0.1 μg of APC-conjugated anti-human CD11b (eBioscience Inc., 17-0118, clone ICRF44) and incubated on ice for 30 min, before washing and formaldehyde fixation. Using a BD FACSCanto II flow cytometer (BD Biosciences), neutrophils were identified by their characteristic forward scatter (FSC) and side scatter (SSC) properties. The corresponding population was gated and the fluorescence analyzed in the APC channel (Ex 635 nm, filter 660/20) to evaluate CD11b expression levels, which were expressed as Mean fluorescence intensity (MFI). Experiments were also carried out using fMLFF concentration curves in the presence of multiple concentrations of antibody to determine the affinity of Fpro0165 for human FPR1 and cynomolgus FPR1 by pA2 (Schild) analysis. Appropriate isotype control IgG were included in all assays.
Measurement of Frpo0165 specificity by FACS
Specificity of the antibody Fpro0165 for FPR1 was determined by assessing potential cross reactivity to its most closely related sequence, FPR2. FACS analysis was carried out using Fpro0165 IgG, and a mouse and a mouse anti-human FPR2 antibody (unpublished in-house reagent) anti-human CXCR3 antibody (R&D Systems, MAB160) with detection using either a PE-labeled anti-human IgG (Sigma P8047) or a PE-labeled anti mouse IgG1(Jackson ImmunoResearch,115-116-146) as appropriate. Isotype controls were a commercially available mouse IgG1 (R&D Systems, Mab 002, clone 11711) or an in-house human IgG1 antibody.
Crystallography and structural determination of Fpro0165 Fab
The Fpro0165 Fab was concentrated to 18.9 mg/ml and crystallized at 20°C using the vapor diffusion method in sitting drops. Drops were set up by mixing equal volumes of Fpro0165 Fab and reservoir solution (0.1 M Tris-HCl pH 7.5-8.0, 18% PEG8000, 0.18-0.2 M MgCl2). The crystals were equilibrated in reservoir solution containing 30% PEG400 and flash frozen in liquid nitrogen. Data were collected at beam line ID23 at the European Synchrotron Radiation Facility. The data were processed, scaled and further reduced using the AutoProc workflow.Citation35 Initial phasing was done by molecular replacement using a high resolution Fab structure (PDB code 1AQKCitation36) as a starting model. The asymmetric unit contained a single Fab molecule, resulting in a solvent content of 71%. Although the data were anisotropic, extending to 3.1 Å in the l direction and 3.8 Å in the orthogonal plane, the resulting electron density maps allowed unambiguous modeling of the backbone and most side chains. Model rebuilding was performed using CootCitation37 and refinement was performed using autobusterCitation38 with LSSR restraints.Citation39 High resolution Fab fragment structures with high homology to Fpro0165 Fab were used to build a model for target restraints (PDB code 3AAZCitation40 and PDB code 3GHBCitation41 for the light and heavy chains respectively). The structural coordinates of Fpro0165 Fab have been deposited in the Protein Data Bank (PDB code 4UV4).
Homology modeling and docking of Fpro0165 Fvand human FPR1
A structural model of human FPR1 (residues S19-P317) was generated using the structure of CXCR4 (PDB code 3OE0)Citation42 as template since both receptors belong to the γ branch of the GPCR superfamily. In order to model the C-terminal helix of FPR1 part of C-terminal sequence from kappa opioid receptor (PDB code 4DJH)Citation43 was also included in the alignment (Fig. S1). Sequences were aligned using Discovery Studio software tools from Accelrys Ltd and further manually edited. The aligned regions of FPR1 and CXCR4 show 28.4% sequence identity and 49.0% sequence similarity. 1000 initial FPR1 models were generated using Discovery Studio tools. The top 10 models were further analyzed and one of these was chosen as the final human FPR1 model. The Ramachandran statistics for this model are 97.9% of the residues are found in the allowed region of the Ramachandran plot and 2.1% in the marginally allowed region. This FPR1 model and the Fv region of the Fpro0165 Fab crystal structure were used in a protein-protein docking protocol to generate a model of the antibody-receptor complex using Discovery Studio tools. Initially 54,000 docking poses were generated and with further analysis a final model was chosen for the complex. Figures were generated using PyMol Molecular Graphics System, Version 1.6.0.0 Schrodinger, LLC.
Data analysis
All data are expressed as mean ± SEM. Data were analyzed using GraphPad PRISM 5.00 for Windows (GraphPad Software Inc.). Where appropriate, IC50 values were determined for each individual experiment and are shown as geometric mean (with 95% confidence limits).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary_Figures_S1-S2.zip
Download Zip (142 KB)Acknowledgments
We thank the MedImmune DNA Chemistry team for antibody DNA sequencing and the MedImmune Biologics Expression and Early Material Supply Teams for antibody protein production. We also thank the MedImmune Core Tissue Culture team for the maintenance and supply of cell lines used in this study.
Funding
JKH was supported by NanoMem Marie Curie Initial Training Network funded by the European Commission within the 7th Framework Programme (project number 317079).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International union of basic and clinical pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev 2009; 61(2):119-61; PMID:19498085; http://dx.doi.org/10.1124/pr.109.001578
- Boulay F, Tardif M, Brouchon L, Vignais P. Synthesis and use of a novel N-formyl peptide derivative to isolate a human N-formyl peptide receptor cDNA. Biochem Biophys Res Commun 1990; 168(3):1103-9; PMID:2161213; http://dx.doi.org/10.1016/0006-291X(90)91143-G
- Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev 2006; 17(6):501-19; PMID:17084101; http://dx.doi.org/10.1016/j.cytogfr.2006.09.009
- Li Y, Ye D. Molecular biology for formyl peptide receptors in human diseases. J Mol Med (Berl) 2013; 91(7): 781-9; PMID:23404331; http://dx.doi.org/10.1007/s00109-013-1005-5
- Zhang Q, Raoof M, Chen Y, Sumi Y, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464(7285): 104-7; PMID:20203610; http://dx.doi.org/10.1038/nature08780
- Liu M, Zhao J, Chen K, Bian X, Wang C, Shi Y, Wang JM. G protein-coupled receptor FPR1 as a pharmacologic target in inflammation and human glioblastoma. Int Immunopharmacol 2012; 14(3):283-8; PMID:22863814; http://dx.doi.org/10.1016/j.intimp.2012.07.015
- Webb DR, Handel TM, Kretz-Rommel A, Stevens RC. Opportunities for functional selectivity in GPCR antibodies. Biochem Pharmacol 2013; 85(2): 147-52; PMID:22975405; http://dx.doi.org/10.1016/j.bcp.2012.08.021
- Niebauer RT, White JF, Fei Z, Grisshammer R. Characterization of monoclonal antibodies directed against the rat neurotensin receptor NTS1. J Recept Signal Transduct Res 2006; 26(5-6):395-415; PMID:17118789; http://dx.doi.org/10.1080/10799890600928228
- Mancia F, Brenner-Morton S, Siegel R, Assur Z, Sun Y, Schieren I, Mendelsohn M, Axel R, Hendrickson WA. Production and characterization of monoclonal antibodies sensitive to conformation in the 5HT2c serotonin receptor. Proc Natl Acad Sci U S A. 2007; 104(11):4303-8; PMID:17360519; http://dx.doi.org/10.1073/pnas.0700301104
- Hutchings CJ, Koglin M, Marshall FH. Therapeutic antibodies directed at G protein-coupled receptors. MAbs 2010; 2(6):594-606; PMID:20864805; http://dx.doi.org/10.4161/mabs.2.6.13420
- Chen CR, McLachlan SM, Rapoport B. Suppression of thyrotropin receptor constitutive activity by a monoclonal antibody with inverse agonist activity. Endocrinology 2007; 148(5):2375-82; PMID:17272389; http://dx.doi.org/10.1210/en.2006-1754
- Tohidkia MR, Asadi F, Barar J, Omidi Y. Selection of potential therapeutic human single-chain fv antibodies against cholecystokinin-B/gastrin receptor by phage display technology. BioDrugs 2013; 27(1):55-67; PMID:23344946; http://dx.doi.org/10.1007/s40259-012-0007-0
- Ravn P, Madhurantakam C, Kunze S, Matthews E, Priest C, O’brien S, Collinson A, Papwort M, Fritsch-Fredin M, Benthem L, et al. Structural and pharmacological characterisation of novel potent and selective monoclonal antibody antagonists of glucose-dependent insulinotropic polypeptide receptor. J Biol Chem 2013; 288(27): 19760-72; PMID:23689510; http://dx.doi.org/10.1074/jbc.M112.426288
- Allard B, Priam F, Deshayes F, Ducancel F, Boquet D, Wijkhuisen A, Couraud JY. Electroporation-aided DNA immunization generates polyclonal antibodies against the native conformation of human endothelin B receptor. DNA Cell Biol 2011; 30(9):727-37; PMID:21688998; http://dx.doi.org/10.1089/dna.2011.1239
- McKnight A, Wilkinson D, Simmons G, Talbot S, Picard L, Ahuja M, Marsh M, Hoxie JA, Clapham PR. Inhibition of human immunodeficiency virus fusion by a monoclonal antibody to a coreceptor (CXCR4) is both cell type and virus strain dependent. J Virol 1997; 71(2):1692-6; PMID:8995702
- Goetzl EJ, Dembrow D, Van Brocklyn JR, Graler M, Huang MC. An IgM-kappa rat monoclonal antibody specific for the type 1 sphingosine 1-phosphate G protein-coupled receptor with antagonist and agonist activities. Immunol Lett 2004; 93(1):63-9; PMID:15134901; http://dx.doi.org/10.1016/j.imlet.2004.02.007
- Ji C, Brandt M, Dioszegi M, Jekle A, Schwoerer S, Challand S, Zhang J, Chen Y, Zautke L, Achhammer G et al. Novel CCR5 monoclonal antibodies with potent and broad-spectrum anti-HIV activities. Antiviral Res 2007; 74(2): 125-37; PMID:17166600; http://dx.doi.org/10.1016/j.antiviral.2006.11.003
- Hagemann UB, Gunnarsson L, Geraudie S, Scheffer U, Griep RA, Reiersen H, Duncan AR, Kiprijanov SM. Fully human antagonistic antibodies against CCR4 potently inhibit cell signaling and chemotaxis. Plos One 2014;9(7) e103776; PMID:25080123; http://dx.doi.org/10.1371/journal.pone.0103776
- Minter RR, Cohen ES, Wang B, Liang M, Vainshtein I, Rees G, Eghobamien L, Harrison P, Sims DA, Matthews C et al. Protein engineering and preclinical development of a GM-CSF receptor antibody for the treatment of rheumatoid arthritis. Br J Pharmacol 2013; 168(1):200-11; PMID:22913645; http://dx.doi.org/10.1111/j.1476-5381.2012.02173.x
- Lowe D, Wilkinson T, Vaughan TJ. Affinity maturation approaches for antibody lead optimization. In: Wood CR, editor. Antibody Drug Discovery. London, UK: Imperial College Press; 2012. p. 85-120.
- Vargas HM, Amouzadeh HR, Engwall MJ. Nonclinical strategy considerations for safety pharmacology: evaluation of biopharmaceuticals. Expert Opinion on Drug Safety 2013; 12(1):91-102; PMID:23170873; http://dx.doi.org/10.1517/14740338.2013.745851
- Kabat EA, Wu TT. Identical V region amino acid sequences and segments of sequences in antibodies of different specificities. Relative contributions of VH and VL gene, minigenes, and complementarity-determining regions to binding of antibody-combining sites. J Immunol 1991; 147:1709-19; PMID:1908882
- Zemlin M, Klinger M, Link J, Zemlin C, Bauer K, Engler JA, Schroder HW Jr, Kirkham PM. Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. J Mol Biol 2003; 334(4):733-49; PMID:14636599; http://dx.doi.org/10.1016/j.jmb.2003.10.007
- Collis AV, Brouwer AP, Martin AC. Analysis of the antigen combining site: correlations between length and sequence composition of the hypervariable loops and the nature of the antigen. J Mol Biol 2003; 325(2): 337-354; PMID:12488099; http://dx.doi.org/10.1016/S0022-2836(02)01222-6
- Dhillon AK, Stanfield RL, Gorny MK, Williams C, Zolla-Pazner S, Wilson, IA. Structure determination of an anti-HIV-1 Fab 447-52D-peptide complex from an epitaxially twinned data set. Acta Crystallogr D Biol Crystallogr. 2008; 64(Pt 7):792-802; PMID:8566514; http://dx.doi.org/10.1107/S0907444908013978
- Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK. Adrenaline-activated structure of beta-adrenoceptor stabilized by an engineered nanobody. Nature 2013; 502(7472):575-9; PMID:24056936; http://dx.doi.org/10.1038/nature12572
- Tillotson BJ, de Larrinoa IF, Skinner CA, Klavas DM, Shusta EV. Antibody affinity maturation using yeast display with detergent-solubilized membrane proteins as antigen sources. Protein Eng Des Sel 2013; 26(2): 101-12; PMID:23109730; http://dx.doi.org/10.1093/protein/gzs077
- Mirzabekov T, Kontos H, Farzan M, Marasco W, Sodroski J. Paramagnetic proteoliposomes containing a pure, native, and oriented seven-transmembrane segment protein, CCR5. Nat Biotechnol 2000; 18(6): 649-654; PMID:10835604; http://dx.doi.org/10.1038/76501
- Hutchings CJ, Cseke G, Osborne G, Woolard J, Zhukov A, Koglin M, Jazayeri A, Pandya-Pathak J, Langmead CJ, Hill SJ et al. Monoclonal anti-beta 1-adrenergic receptor antibodies activate G protein signaling in the absence of beta-arrestin recruitment. MAbs 2014; 6(1):246-61; PMID:24253107; http://dx.doi.org/10.4161/mabs.27226
- Sahagun-Ruiz A, Colla JS, Juhn J, Gao JL, Murphy PM, McDermott DH. Contrasting evolution of the human leukocyte N-formylpeptide receptor subtypes FPR and FPRL1R. Genes Immun 2001; 2(6):335-42; PMID:11607790; http://dx.doi.org/10.1038/sj.gene.6363787
- Kilpatrick KE, Wring SA, Walker DH, Macklin MD, Payne JA, Su JL, Champion BR, Catterson B, McIntyre GD. Rapid development of affinity matured monoclonal antibodies using RIMMS. Hybridoma 1997; 16(4): 381-9; PMID:9309429; http://dx.doi.org/10.1089/hyb.1997.16.381
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256(5517):495-7; PMID:1172191; http://dx.doi.org/10.1038/256495a0
- Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol 1987; 154:367-82; PMID:3323813; http://dx.doi.org/10.1016/0076-6879(87)54085-X
- Oganesyan V, Gao C, Shirinian L, Wu H, Dall'Acqua WF. Structural characterization of a human fc fragment engineered for lack of effector functions. Acta Crystallogr D Biol Crystallogr. 2008; 64(Pt 6): 700-4; PMID:18560159
- Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr 2011; 67(Pt 4): 293-302; PMID:21460447; http://dx.doi.org/10.1107/S0907444911007773
- Faber C, Shan L, Fan Z, Guddat LW, Furebring C, Ohlin M, Borrbaeck CA, Edmundson AB. Three-dimensional structure of a human fab with high affinity for tetanus toxoid. Immunotechnology 1998; 3(4): 253-70; PMID:9530559; http://dx.doi.org/10.1016/S1380-2933(97)10003-3
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D Biol Crystallogr 2010; 66(Pt 4):486-501; PMID:20383002; http://dx.doi.org/10.1107/S0907444910007493
- Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C et al. 2011. BUSTER version 2.11.5. Cambridge, UK: Global Phasing Ltd.
- Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr 2012; 68(Pt 4): 368-80; PMID:22505257; http://dx.doi.org/10.1107/S0907444911056058
- Robert R, Streltsov VA, Newman J, Pearce LA, Wark KL, Dolezal O. Germline humanization of a murine abeta antibody and crystal structure of the humanized recombinant fab fragment. Protein Sci 2010; 19(2): 299-308; PMID:20014445; http://dx.doi.org/10.1002/pro.312
- Burke V, Williams C, Sukumaran M, Kim SS, Li H, Wang XH, Gorny MK, Zollar-Pazner S, Kong XP. Structural basis of the cross-reactivity of genetically related human anti-HIV-1 mAbs: implications for design of V3-based immunogens. Structure 2009; 17(11): 1538-46; PMID:19913488; http://dx.doi.org/10.1016/j.str.2009.09.012
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010; 330(6007): 1066-71; PMID:20929726; http://dx.doi.org/10.1126/science.1194396
- Wu H, Wacker D, Mileni M, Katritch V, Won Han G, Vardy E, Liu W, Thompson AA, Huang X, Carroll RI et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012; 485(7398): 327-32; PMID:22437504; http://dx.doi.org/10.1038/nature10939