Abstract
Mitochondrial health is maintained by the quality control mechanisms of mitochondrial dynamics (fission and fusion) and mitophagy. Decline of these processes is thought to contribute to aging and neurodegenerative diseases. To investigate the role of mitochondrial quality control in aging on the cellular level, human umbilical vein endothelial cells (HUVEC) were subjected to mitochondria-targeted damage by combining staining of mitochondria and irradiation. This treatment induced a short boost of reactive oxygen species, which resulted in transient fragmentation of mitochondria followed by mitophagy, while mitochondrial dynamics were impaired. Furthermore, targeted mitochondrial damage upregulated autophagy factors LC3B, ATG5 and ATG12. Consequently these proteins were overexpressed in HUVEC as an in vitro aging model, which significantly enhanced the replicative life span up to 150% and the number of population doublings up to 200%, whereas overexpression of LAMP-1 did not alter the life span. Overexpression of LC3B, ATG5 and ATG12 resulted in an improved mitochondrial membrane potential, enhanced ATP production and generated anti-apoptotic effects, while ROS levels remained unchanged and the amount of oxidized proteins increased. Taken together, these data relate LC3B, ATG5 and ATG12 to mitochondrial quality control after oxidative damage, and to cellular longevity.
Introduction
Mitochondrial quality control (QC) mechanisms encompass the removal of damaged mitochondrial components by the mitochondrial lon protease, mitochondrial dynamics (fusion and fission) and whole organelle degradation (mitophagy), thus ensuring the maintenance of a healthy mitochondrial population.Citation1,Citation2 Failure of mitochondrial QC is proposed to participate in the development and progression of Alzheimer and Parkinson diseases as well as in the aging process. While mitochondrial QC is an ongoing process, it becomes specifically important in the management of reactive oxygen species (ROS)-induced damage, as ROS are produced by the mitochondrial respiratory complexes I and III and affect primarily mitochondrial DNA (mtDNA), lipids and proteins.
After oxidative damage, mitochondria can either be degraded via mitophagy or rescued by mitochondrial dynamics. mtDNA and mitochondrial proteins including the complexes of the respiratory chain can be exchanged by fusion and fission.Citation3–Citation6 An intact membrane potential is necessary for the fusion of the inner mitochondrial membrane, and thus the exchange of inner membrane proteins and matrix components.Citation7 We actually observed a decline of mitochondrial dynamics after addition of hydrogen peroxide,Citation8 implying that mitochondrial dynamics may not be the primary QC mechanism utilized after oxidative stress.
Therefore, it seems more likely that a damaged mitochondrion or a mitochondrial segment separated by fission will be degraded by mitophagy.Citation9 The signal that initiates autophagy of a damaged mitochondrion is still unclear; however, a loss of mitochondrial membrane potential and mitochondrial fragmentationCitation9,Citation10 as well as translocation of the PARK2-associated ubiquitin ligase Parkin from the cytosol to the mitochondrionCitation11,Citation12 are involved in the initiation of the autophagic process. During mitophagy the dysfunctional mitochondrion is engulfed by a membranous structure, the phagophore, which expands into an autophagosome. After fusion with endosomes and/or lysosomes the contents of the autolysosome are degraded.
The proteins of the ATG family regulate autophagy. ATG5, ATG12 and LC3B (ATG8F) all participate in the formation of the autophagosome. ATG5 forms a complex with ATG12 and ATG16L at the membrane of the evolving autophagosome.Citation13 The 18-kDa isoform LC3-I is localized in the cytosol and after conjugation to phosphatidylethanolamine the activated 16-kDa isoform LC3-II is situated within the autophagosomal membrane.Citation14 The lysosomal protein LAMP-1 is a glycoprotein that colocalizes with lysosomal staining and in cooperation with LAMP-2 participates in the autophago-lysosomal pathway.Citation15 Furthermore, LAMP-1 and LAMP-2 are thought to mediate fusion of phagosomes with late endosomes.Citation16
According to the “garbage theory of aging,” an accumulation of ROS-damaged molecules mediated by dysfunctional QC contributes to aging.Citation17 This theory is supported by a decrease of mitochondrial dynamics in senescent cells,Citation18,Citation19 reduced expression of different autophagosomal proteins in old D. melanogasterCitation20 and decrease of the lysosomal protein LAMP-2A in the livers of old rats.Citation21
Caloric restriction and insulin resistance extend the life span of different model systems,Citation22–Citation25 and enhanced autophagy might be one of the main reasons behind the increased life spans.Citation26–Citation28 This hypothesis correlates with data that demonstrate increased life span of D. melanogaster after overexpression of ATG8ACitation20 and extended life span of yeast after treatment with the autophagy-inducing drug rapamycin.Citation29 In contrast, some autophagy genes such as the ATG1 homolog ULK3 are upregulated in senescent human diploid fibroblasts, and overexpression of ULK3 results in premature senescence of these cells, accordingly implying a role of ULK3 in the mediation of senescence.Citation30
Taken together, the role of mitochondrial QC and of the different autophagy genes after ROS damage and in the aging process are still unclear. Therefore, we analyzed the QC mechanisms after oxidative stress and the role of the involved ATG proteins ATG5, ATG12 and LC3B in regard to mitochondrial fitness and life span.
Results
Mitochondrial dynamics are not involved in early QC after ROS-induced mitochondrial damage in young HUVEC.
According to the mitochondrial free-radical hypothesis of aging,Citation31 a vicious circle of mitochondrial ROS production and mitochondrial dysfunction participates in the aging process or even is the main cause of cellular aging. The QC mechanisms of mitochondrial dynamics and mitophagy are hypothesized to maintain mitochondrial fitness to counteract ROS-induced damage.Citation1 To test this hypothesis young human umbilical vein endothelial cells (HUVEC) were subjected to general and mitochondria-targeted oxidative stress. General oxidative stress was achieved by short-term exposure to hydrogen peroxide, while mitochondrial damage was inflicted by a phototoxicity method where mitochondria were stained with the photoactive dye MitoTrackerRed (MTR) and subsequently irradiated with green light. Both methods result in a transient ROS boost as shown previously.Citation8,Citation19 Quantification of oxidized proteins immediately after stress exposure demonstrated that mitochondria-targeted irradiation caused preferential mitochondrial damage, whereas hydrogen peroxide damaged predominantly cytosolic proteins ().
Mitochondrial damage of irradiated cells was further indicated by mitochondrial fragmentation () and loss of membrane potential 4 h after irradiation,Citation19 while stained mitochondria of nonirradiated cells retained their tubular morphotype. To determine the role of mitochondrial dynamics in QC after irradiation, HeLa cells were transfected with mitochondria-targeted CFP (mitoCFP). After 48 h, mitoCFP-expressing cells were stained with MTR, either irradiated or nonirradiated and then co-seeded with nonirradiated cells transfected with mitoGFP. After 4 h, cells were fused by addition of polyethyleneglycol (PEG). Mitochondria of nonirradiated control cells engaged in mitochondrial fusion and fission as evidenced by the mixing of the dyes CFP/MTR and GFP ( and C). In contrast, CFP/MTR and GFP did not mix in irradiated cells, neither 4 h nor 24 h after irradiation ( and C), indicating that damaged mitochondria as characterized by fragmentation and a strong loss of membrane potential are not repaired by mitochondrial dynamics in the first 24 h after damage.
Mitophagy is the primary QC mechanism after ROS-induced mitochondrial damage in young HUVEC.
As fusion and fission were not active up to 24 h after irradiation, we investigated the putative clearance of dysfunctional mitochondria by the autophagolysosomal pathway. The Parkinson disease-associated protein Parkin (an E3 ubiquitin ligase) is recruited to mitochondria 2 h after CCCP-induced loss of membrane potential.Citation11 As irradiation-damaged mitochondria exhibit a reduced membrane potential,Citation19 young HUVEC were transfected with GFP-Parkin, stained with MTR after 2 d, irradiated and analyzed by CLSM. Indeed, 2 h after irradiation Parkin had translocated from the cytosol to the fragmented mitochondria, implying tagging of mitochondria for degradation (). Quantification of LC3-II after irradiation by western blotting showed a time-dependent increase of LC3-II normalized to actin with a peak at 24 h after irradiation, supporting the induction of mitophagy by irradiation ( and C).
Damaged MTR-stained mitochondria without intact membrane potential started to appear in perinuclear localized clusters 24 h post irradiation, discernable from mitochondria with intact membrane potential by the lack of MitoTracker Green signal after re-staining (). The amount of mitochondrial clusters increased up to 72 h after irradiation before it started to decrease (). In parallel we observed an increase of the lysosomal area visualized by LAMP-2 staining until 72 h after irradiation ( and G). In correlation with these data a phasal increase of the LAMP-2 protein with a peak from 24 h to 48 h after irradiation became apparent ( and I). When irradiated and hydrogen peroxide-treated cells were analyzed by electron microscopy autophagosomes with mitochondrial content (arrow) appeared preferentially in irradiated cells but not in nontreated cells (Fig. S1). The induction of mitophagy after irradiation was further supported by colocalization of fragmented mitochondria with LC3-containing vesicles (Fig. S2) and LAMP-2-containing vesicles (Fig. S3) in irradiated cells. Taken together, these data indicate clearly that mitophagy acts as QC mechanism after irradiation, whereupon damaged mitochondria are transported to the perinuclear region, then engulfed by autophagosomes and finally degraded in lysosomes.
Overexpression of ATG5, LC3B and ATG12 increase life span of two cell models.
The data presented in and indicate that removal of damaged mitochondria by mitophagy is an early step to counteract oxidative damage. To determine which factors of the autophagy pathway were activated after irradiation, the mRNA expression of various autophagy genes was analyzed by semiquantitative RT-PCR 8 h and 72 h after irradiation. A significant increase of the ATG5, LC3B and ATG12 mRNAs was observed at both time points, while ATG7 and ATG10 showed no induction (). In parallel, a strong increase of the PGC1α mRNA occurred, indicating de novo synthesis of mitochondrial components to replace degraded mitochondria.
To confirm our hypothesis that these upregulated autophagy factors enhance mitophagy/mitochondrial turnover we first stained mitochondria of HeLa cells with MTR, which binds covalently to mitochondrial proteins, and then transfected these cells transiently with plasmids expressing either GFP-LC3B, GFP-ATG5 or GFP as a control. Forty-eight hours later the relative MTR fluorescence was measured and a significant decline of the MTR fluorescence in the GFP-LC3B and GFP-ATG5 cells in comparison to GFP transfected cells became apparent (), indicating that overexpression of GFP-LC3B and GFP-ATG5 indeed increased mitophagy.
Based on these data we hypothesized that increased mitophagy throughout the replicative life span may be beneficial for mitochondrial and cellular fitness. Previously, we showed that over-expression of gATG5 extended the replicative live span of chicken embryonic fibroblasts (CEF), an avian in vitro aging model.Citation32 Therefore, the human autophagy proteins LC3B, ATG5 and ATG12 were stably overexpressed as GFP fusion proteins in HUVEC as a human in vitro aging model. For gene transfer the retroviral replication-competent avian leukosis virus long-terminal repeat, splice acceptor (RCAS)-A virus system and young HUVEC stably overexpressing the RCAS-A virus receptor (TVA receptor) were used (see Material and Methods). These HUVEC had passed 13 population doublings (PD) at the time point of virus infection. As controls, HUVEC with TVA receptor but without virus infection (“nontransfected”) and GFP-overexpressing cells were used as well as cells overexpressing the lysosomal protein LAMP-1-GFP.
Overexpression of GFP-LC3B in young HUVEC resulted in a diffuse cytosolic GFP fluorescence and strongly fluorescent spots, representing the LC3-I and LC3-II isoforms, respectively, whereas LAMP-1-transfected cells revealed a large population of lysosomes (). In old GFP-LC3B transfected cells LC3Bpositive autophagosomes were more numerous and larger than in young ones. GFP-ATG5-transfected HUVEC displayed GFP-stained autophagosomes only in old HUVEC, while young cells exhibited only cytoplasmatic staining in accordance with previous recordsCitation33,Citation34 and GFP-ATG12-overexpressing cells looked similar (). Overexpression of the full-length recombinant proteins was confirmed by western blotting with antibodies against GFP, LC3, ATG5 and LAMP-1 respectively (Fig. S4).
Young TVA-expressing HUVEC at PD 13 were infected with RCAS virus coding for GFP, GFP-ATG5, GFP-LC3B, GFP-ATG12 and LAMP-1-GFP or noninfected (“nontransfected”) (depicted as PD 0 in ) and were cultivated in triplicate until they reached replicative senescence. Overexpression of the autophagy proteins ATG5, LC3B and ATG12 extended the life span of HUVEC significantly compared with nontransfected and GFP-transfected cells. In contrast, cells overexpressing the lysosomal protein LAMP-1 exhibited a replicative life span similar to both control cell populations. Control cells and LAMP-1-overexpressing cells reached replicative senescence already after 60 d and around 10 PD (in total 23 PD), while LC3B, ATG5 and ATG12 overexpression extended the replicative life span of HUVEC up to 90 d and 22 PD (in total 35 PD) ().
The life-span extending effect was confirmed in a second in vitro aging model. Young CEF were infected with RCAS virus coding for GFP and GFP-gLC3B at passage 8 (depicted as PD 0 in ) and were cultivated in triplicate until they reached replicative senescence. In CEF, the life-span extension was less pronounced but still significant; overexpression of gLC3B increased the PD number to 42 PD compared with GFP-expressing cells that reached 35 PD (). As with LC3B-, ATG5-and ATG12-overexpressing HUVEC the life span extending effect took place in the late stage of the growth curve.
Therefore, the effect of LC3B, ATG5 and ATG12 over-expression was investigated in pre-senescent cells. TVA-expressing HUVEC at PD 22 were stably transfected with GFP, LC3B, ATG5 and ATG12 or not infected (“nontransfected”). After an additional 30 d in culture no difference between transfected and nontransfected cells was discernable (). Taken together, these data indicate that overexpression of the autophagy proteins LC3B, ATG5 and ATG12 prolonged life span and increased the PD number of two in vitro aging models but only if the proteins were already overexpressed in young cells.
Overexpression of autophagy factors enhances cellular and mitochondrial fitness.
Autophagy and apoptosis can regulate and influence each other, and HUVEC as well as CEF populations exhibit increased apoptosis rates in replicative senescence.Citation32,Citation35 Therefore, the observed life-span enhancing effect after over-expression of autophagy proteins may be at least partially mediated by reduced apoptosis especially in the later stages of the growth curve. To test this hypothesis HUVEC overexpressing LC3B, ATG5, ATG12 or LAMP-1, as well as CEF overexpressing gLC3B were subjected to short-term exogenous oxidative stress. Overexpression of autophagy proteins or LAMP-1 rendered cells more resistant against induction of hydrogen peroxide-induced apoptosis compared with nontransfected controls or GFP over-expressing cells ( and B).
According to the mitochondrial hypothesis of agingCitation31 an extended life span should correlate with enhanced mitochondrial fitness. Mitochondrial membrane potential is a good marker for mitochondrial fitness and therefore was evaluated by staining young and senescent HUVEC and CEF overexpressing ATG5, LC3B, ATG12 or LAMP-1 with the mitochondrial membrane potential indicator TMRE. Quantification of the TMRE fluorescence revealed that the membrane potential was improved in young cells overexpressing the autophagy proteins or LAMP-1 in comparison to control cells (nontransfected and GFP overexpressing cells), whereas overexpression of ATG5, ATG12 and LC3B had a stronger effect than overexpression of LAMP-1 ( and D). In senescent cells, however, enhanced expression of autophagy proteins did not affect the membrane potential (Fig. S5A and S5B), indicating again an age-dependent protective effect of the overexpressed proteins. A higher membrane potential can be assumed to result in increased ATP production. Therefore, the ATP content in young ATG5-, LC3B-, ATG12-and LAMP-1-overexpressing HUVEC was measured and compared with GFP-transfected cells (). All autophagy protein over-expressing cells contained significantly more ATP than LAMP-1- or GFP-expressing cells, supporting the data of an improved membrane potential.
As an additional marker for mitochondrial fitness, the integrity of the mtDNA was determined. For this purpose, total DNA from nontransfected and GFP-LC3B-overexpressing HUVEC was isolated at different time points of the growth curve and a small (0.2 kb) and a large (8.9 kb) fragment of the mtDNA were amplified by semiquantitative PCR; the small fragment was used for normalization. The relative amplification of the 8.9-kb fragment acts as parameter for mtDNA integrity and mtDNA damage.Citation18 In LC3B-overexpressing cells the relative amplification of the 8.9-kb fragment was significantly stronger than in nontransfected cells (), which becomes especially clear when comparing presenescent cells (50–60 d after infection), indicating reduced mtDNA damage due to the LC3 overexpression.
Taken together, overexpression of ATG5, LC3B, ATG12, and, to a lesser amount, overexpression of LAMP-1, confers cell-protective effects, which correlate well with the extended life span.
Overexpression of LC3B, ATG5 and ATG12 results in an increase of oxidized proteins.
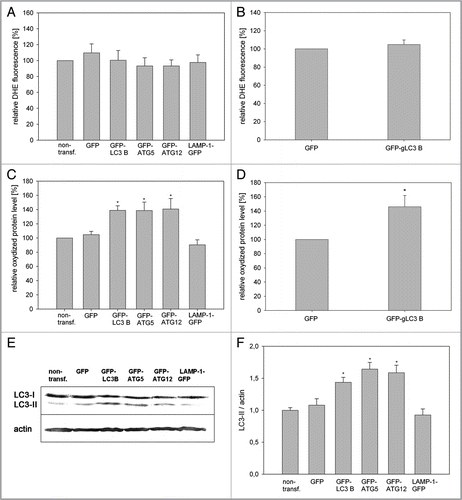
According to the mitochondrial hypothesis of aging,Citation31 cells with an extended life span would be expected to have a lower oxidative load. Thus, the ROS content of young and old LC3B-, ATG5-, ATG12-and LAMP-1-overexpressing HUVEC, and gLC3B-overexpressing CEF was determined by staining cells with the ROS indicator DHE. Interestingly, none of these proteins influenced ROS levels, neither in HUVEC nor in CEF, whether young or senescent ( and B; Fig. S6A and S6B).
The lack of influence of autophagy protein overexpression on ROS levels seemingly stands in contrast to their effect on life span and cellular and mitochondrial fitness. As ROS measurements only reveal the actual amount of free radicals, whereas the amount of oxidized proteins can be considered a crucial parameter representing ROS load, the carbonylation status of cellular proteins was determined by oxyblotting. Whole-cell lysates of young and senescent gLC3B and GFP CEF, ATG5, ATG12, LC3B, LAMP-1 and GFP HUVEC were separated by gel electrophoresis and the amount of carbonylated proteins was quantified. Interestingly, overexpression of the autophagy proteins in young HUVEC and young CEF resulted in a significantly increased amount of oxidized proteins compared with the controls (GFP and non-transfected) and LAMP-1 transfected cells ( and D, and Fig. S7). In old HUVEC overexpressing autophagy proteins the amount of oxidized proteins was slightly but not significantly increased compared with GFP-expressing cells, whereas no difference was found between old GFP and GFPgLC3B CEF (Fig. S8A and S8B).
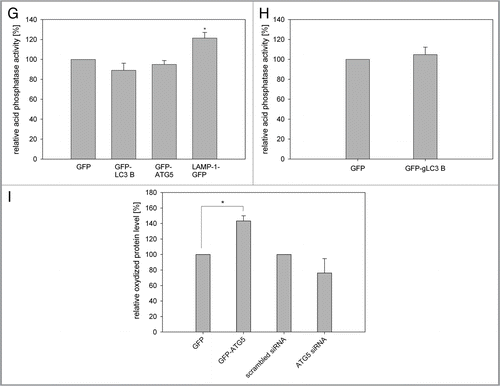
A possible explanation of these data would be a storage of oxidized proteins and damaged mitochondria in LC3B-, ATG5- and ATG12-overexpressing cells, most probably in autophagosomes, as overexpression of LAMP-1 neither extended the life span or induced an increase of oxidized proteins. This hypothesis was tested in young HUVEC by determining the ratio of the endogenous, autophagosome-bound LC3-II vs. actin. Stable LC3B-, ATG5-and ATG12-overexpressing HUVEC had a much higher LC3-II/actin ratio than control cells (GFP and nontransfected cells) or LAMP-1 overexpressing cells ( and F). Even after a transient transfection of young HUVEC with LC3B, ATG5 and ATG12 a significant increase of the LC3-II/actin ratio of LC3B and ATG5 transfected cells compared with the controls was observed (Fig. S9). For further confirmation we analyzed the lysosomal activity of young LC3B-and ATG5-overexpressing HUVEC and of gLC3B-overexpressing CEF. No significant increase of acid phosphatase activity was visible in cells overexpressing autophagy proteins compared with GFP-overexpressing cells ( and H), indicating that the lysosomal activity was not altered by overexpression of autophagy proteins. In contrast, over-expression of LAMP-1 caused a significant increase of acid phosphatase activity (). Also the lysosomal area was not increased in HUVEC overexpressing LC3B compared with nontransfected cells (Fig. S10), implying together with the data presented above an accumulation of autophagosomes, and thus a stall in autophagic flux due to overexpression of LC3B, ATG5 and ATG12.
In a further experiment we modulated the ATG5 protein content in young HUVEC by transient transfection with GFP-ATG5 and a siRNA against the endogenous ATG5. 72 h after transfection of the ATG5 siRNA the amount of endogenous ATG5 protein was reduced by 60% compared with cells transfected with the scrambled siRNA (Fig. S11). The amount of oxidized proteins 72 h after transfection was significantly higher in GFP-ATG5-expressing cells compared with GFP transfected cells, while HUVEC treated with the ATG5 siRNA exhibited fewer oxidized proteins although not significantly, thus supporting the idea of an accumulation of oxidized proteins and damaged mitochondria in autophagosomes ( and Fig. S12). This as well as the increased resistance against apoptosis possibly contributes to the improved mitochondrial fitness and the prolonged replicative life span of ATG5-, ATG12-and LC3B-overexpressing cells.
Discussion
Mitochondrial QC after oxidative stress.
We analyzed the QC mechanisms after exposure of mitochondria to oxidative stress by using a combination of MTR staining and irradiation to induce ROS production in mitochondria, resulting in mitochondrial fragmentation, decrease of mitochondrial membrane potential and oxidation of proteins. Interestingly, this treatment completely blocked exchange of mitochondrial matrix components for at least 24 h after irradiation, indicating that mitochondrial fusion was inhibited. This could be due to the loss of mitochondrial membrane potential as fusion of the inner mitochondrial membrane requires an intact membrane potential.Citation7 Also, application of general oxidative stress (short-term exposure to exogenous hydrogen peroxide) reduced the membrane potential and impaired mitochondrial dynamics.Citation8
While mitochondrial dynamics are inhibited, damaged mitochondria are tagged by Parkin, followed by mitophagy similar to the situation after CCCP-induced loss of membrane potential in HeLa and neuroblastoma cells.Citation11,Citation36 In parallel, an increase of PGC1α, the main transcription factor for mitochondrial proteins, occurred, indicating the activation of synthesis of antioxidant proteinsCitation37,Citation38 and of new mitochondrial components;Citation39 a similar activation of PGC1α has been evoked by hydrogen peroxide treatment of HUVEC.Citation8 PGC1α-driven mitochondrial biogenesis seems to be a common reaction to transient stress as transient hypoxia also induces PGC1α,Citation40 while PGC1α becomes down-regulated during chronic hypoxia.Citation41 As we observed an increase in mitochondrial dynamics 48 h after hydrogen peroxide treatment,Citation8 we propose that irreversibly ROS-damaged mitochondria are degraded by mitophagy, whereas later, less severely damaged mitochondria are rescued by mitochondrial dynamics and biogenesis.
Increased mitophagy extends life span of two different in vitro aging models.
ATG5 and ATG12 as well as LC3B became also upregulated after photodamage of mitochondria, indicating their involvement in mitophagy after oxidative stress. This result is supported by data, which demonstrate an inhibition of mitophagy after knockdown of ATG5 and LC3Citation42 or disruption of the ATG12-ATG3 complex.Citation43 In accordance with our results, transient overexpression of LC3B and ATG5 caused decreased MTR staining over time, indicating that overexpression of these proteins does indeed increase mitophagy.
We have demonstrated previously that overexpression of the chicken ortholog of ATG5 extended the replicative life span of CEF.Citation32 Therefore, overexpression of the mitophagy-associated proteins ATG5, ATG12 and LC3B was investigated in HUVEC as a human in vitro aging system. Endothelial cells demonstrate in vivo and in vitro a prolonged senescent phase, and senescent endothelial cells probably contribute to the development of arteriosclerosis,Citation44 thus presenting an aging-relevant cell model. All three autophagy proteins, but not the lysosomal protein LAMP-1, extended life span and PD number significantly when overexpressed in HUVEC. This effect was species-overlapping as overexpression of gLC3B and gATG5 also increased the life span of CEF. These results support the hypothesis that increased autophagy can extend the life span,Citation26–Citation28 as shown before on an organismal level. Addition of the autophagy-inducing drug rapamycin extends the life span of a yeast modelCitation29 and a murine model,Citation45 and overexpression of Atg8 in D. melanogaster increases the life span of the fly.Citation20 However, overexpression of an ATG1 homolog (ULK3) causes premature senescence in ras-expressing human diploid fibroblasts,Citation30 indicating a differential role of autophagy proteins in life-span determination and/or cell type-specific effects. In contrast to the data obtained from autophagy geneoverexpressing HUVEC, we saw no effect of LAMP-1 overexpression on the in vitro aging process. Also LAMP-1 knockout mice did not demonstrate premature aging,Citation46 implying that LAMP-1 may be not directly involved in the aging process.
Mechanisms of life-span prolongation mediated by overexpression of autophagy proteins.
The increased life span and PD number of the ATG5-, ATG12-and LC3B-overexpressing cell populations could be partially attributed to a reduced apoptotic rate. Apoptosis of HUVEC increases with ageCitation35,Citation47 and as expression of recombinant autophagy proteins confers increased resistance against oxidative stress, this will probably contribute to life-span extension, especially in the later stages of the growth curve. These results correlate with recent data implying that autophagy and apoptosis may influence one another; reduced autophagic rates result in enhanced apoptosis against different stressors,Citation48–Citation50 whereas increased autophagy reduces the amount of apoptotic cells after application of stress.Citation32,Citation51–Citation53
However, the opposing effects of autophagy and apoptosis cannot be the only factor involved in the observed life-span extension, because LAMP-1 overexpression conveyed increased resistance against hydrogen peroxide to HUVEC similar to overexpression of ATG5, ATG12 and LC3B, but remained ineffective in life-span prolongation. ATG5-, ATG12-and LC3B-overexpressing cells were characterized by an improved membrane potential and increased ATP production, whereas LAMP-1 overexpressing cells demonstrated no changes of the ATP content. According to the mitochondrial hypothesis of agingCitation31 mitochondria are key players in aging, and a vicious circle of mitochondrial ROS production and accordingly mitochondrial damage is proposed to promote aging. mtDNA of mutator miceCitation54,Citation55 contains a high amount of mutations due to a defective mtDNA polymerase, and these animals exhibit reduced respiratory chain complex activity, lower ATP levels and premature aging compared with the control mice. In reverse this would mean that a population of healthy mitochondria could mediate longevity and, indeed, in HUVEC overexpressing autophagy genes different parameters provide evidence for enhanced mitochondrial fitness including improved mtDNA integrity. In contrast, prolonged mtDNA damage after oxidative stress resulted in premature senescence of HUVEC,Citation8 thus supporting a possible positive correlation between intact mtDNA, mitochondrial fitness and aging.
The ROS content in young as well as in old HUVEC and CEF overexpressing autophagy genes was similar to control cells, but, interestingly, the amount of oxidized proteins and autophagosomes was significantly increased in both cell types overexpressing autophagy proteins. Thus, the extended live span of autophagy overexpressing cells cannot be attributed to low ROS levels as the free radical hypothesis of aging would predict.Citation31 The role of oxidative stress in aging is quite complex and appears to depend on the model systems, the applied stressor, the respective overexpressed or downregulated gene and possible compensatory mechanisms. Although senescent HUVEC showed an increase of carbonylated proteins,Citation35 oxidized proteins seemingly do not affect cellular fitness and live span in autophagy genes overexpressing HUVEC. A possible explanation would be the storage of damaged mitochondria and oxidized proteins in autophagosomes. This hypothesis is supported by our data showing a correlative modulation of oxidized proteins after alteration of ATG5 protein levels.
Taken together, overexpression of ATG5, ATG12 and LC3B resulted in a healthy mitochondrial population and an extended life span in two different in vitro model systems, stressing the important role of mitophagy as a cell-protective mechanism.
Material and Methods
Cultivation of cells.
HUVEC: Cells in passage 0 were purchased from Promocell (C-12200) and cultivated in Endothelial Cell Growth Medium (Promocell, C-22010) in flasks that had been coated with 0.2% gelatin (Sigma, G1393) at 37°C. To minimize the influence of genetic factors, HUVEC from at least three different isolations were used for the experiments. In growth curves, PD were determined by the following equation: PD = 3.32 * (log10 UCY − log10 I) + X (where UCY is the number of cells at the end of the passage; I the number of cells that were seeded at the beginning of the passage and X the previous PD number. The senescent status of aging cells was verified by staining cells with the Senescence Cells Histochemical Staining Kit (Sigma, CS0030) for SA-β-galactosidase.
CEF: Chicken (white leghorn) eggs (fertile specific pathogen free) were obtained from Charles River Laboratories (SPF, Hungary, MO25) and CEF were prepared from 10-d-old embryos as described before.Citation32 Cells were cultivated at 41°C in IMDM (Invitrogen, 21980) with 5% FCS (Invitrogen, 10270-106), 5% chicken serum (Invitrogen, 16110082) and 1% penicillin/streptomycin (Invitrogen, 15140) in flasks coated with 0.2% gelatin (Sigma, G1393).
HeLa: HeLa cells were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ, DSM-No. ACC 57) and were cultured in Minimal Essential Medium with Earle's salts (Invitrogen, 31095), supplemented with 10% FCS (Invitrogen, 10270-106) and 1% MEM nonessential amino acids (Invitrogen, 11140) at 37°C.
Constructs and stable transfection.
The ORF encoding human LC3B (GeneID: 81631) was amplified by RT-PCR from HUVEC cDNA and cloned into the BglII and EcoRI sites of vector EGFP-C1 (Clontech, 6084-1). For GFP-ATG5 the ORF encoding ATG5 (GeneID: 9474) was amplified from HUVEC cDNA and cloned into the XhoI and BamHI sites of EGFP-C1. ATG12 (GeneID, 9140) was also amplified from HUVEC cDNA and cloned into the EcoRI and BamHI sites of EGFP-C1. The ORF of human LAMP-1 (GeneID, 3916) was amplified by RT-PCR from HUVEC cDNA and cloned into the BglII and EcoR1 sites of vector EGFP-N1 (Clontech, 6085-1), resulting in LAMP-1-GFP. The ORF encoding gLC3B (GeneID: 427559) was amplified by RT-PCR from CEF cDNA and cloned into the EcoRI and BamH1 sites of vector EGFP C1. For GFP-Parkin the vector EGFP-C1 was digested with EcoRI and BamHI. The Parkin-encoding ORF was obtained by RT-PCR from HUVEC cDNA and was cloned into these sites. MitoGFP has been described beforeCitation19 and mitoCFP was obtained from Clontech (6903-1). Constructs were verified by sequencing.
For transient knockdown of Atg5, an ATG5 siRNA (Qiagen, Hs_APG5L_3) was used.
HeLa cells were transfected with Effectene (Qiagen, 301425) according to the manufacturer's instructions. HUVEC were transfected by electroporation using the HUVEC transfection kit (Lonza, VPB-1002) and the Nucleofector II (Lonza, AAD-1001) according to the manufacturer's instructions.
Stable transfection was achieved with a modified RCAS A system, a retroviral vector that had been derived from the SR-A strain of Rous sarcoma virus.Citation32,Citation56 The CMV promotor and the ORFs encoding GFP-ATG5, GFP-ATG12, GFP-LC3B, LAMP1-GFP, and GFP respectively, were amplified by PCR and cloned into the ClaI site of the RCAS A vector (for detailed vector information see www.retrovirus.info/RCAS/index.html) resulting in R-C-GFP-LC3B, R-C-GFP-ATG5, R-C-GFP-ATG12, R-C-LAMP-1-GFP and R-C-GFP, respectively.
Plasmids R-C-GFP-LC3B, R-C-GFP-ATG5, R-C-GFP-ATG12, R-C-LAMP-1-GFP and R-C-GFP were transfected with Effectene into young CEF according to the manufacturer's instructions. One week after transfection, almost all CEF showed GFP fluorescence with the respective localization, indicating infection of the whole cell population with recombinant virus. For stable transfection of HUVEC the RCAS-A virus receptor (TVA receptor) was amplified by RT-PCR from CEF cDNA and cloned into the BamHI and NotI sites of the EGFP-N1 vector where the GFP had been deleted. The TVA receptor was transfected into HUVEC by electroporation and stably transfected cells were selected with G418 (Invitrogen, 11811) resistance. Stable TVA HUVEC were exposed for 24 h to 3 d old virus-containing supernatant of infected CEF mixed with endothelial medium (1:1) and expression of the recombinant proteins was confirmed by microscopy and western blotting.
Induction of oxidative stress.
General oxidative stress was induced by subjecting cells to a single dose of hydrogen peroxide (30%, Calbiochem, 386790) in the range from 3.3 mM to 13.2 mM for 10 min in culture medium. After hydrogen peroxide exposure the medium was replaced by fresh culture medium. At the indicated time points the remaining adherent cells were detached with trypsin (Invitrogen, 15090) and counted in a Neubauer chamber to obtain the number of surviving cells.
Irradiation was performed as described earlier.Citation19 Briefly, mitochondria were stained with the photoactive dye MitoTracker Red CMX ROS (MTR) (Invitrogen, M-7512) and irradiated for 15 min with green light with an intensity of 0.3 J/cm2.
Western blotting, oxyblot and isolation of mitochondria.
Protein lysates were separated by SDS-PAGE and transferred by semidry blotting onto a nitrocellulose membrane. GFP and GFP fusion proteins were detected with a polyclonal anti-GFP antibody (Clontech, 632460), endogenous LC3 and ATG5 with the respective polyclonal antibodies (Sigma, L8918 and A0856) and LAMP-2 with a monoclonal antibody (BD Bioscience PharMingen, 555803). For visualization of the bands the NEB/BCIP system (Sigma, B6404) for detection of alkaline phosphatase activity or the SuperSignal® West Pico Chemiluminescent Substrate (Thermo, 34080) for detection of horseradish peroxidase activity was used. The actin levels were determined with a polyclonal anti-actin antibody (Sigma, A5056). The quantification of the respective gray values of all bands was achieved with the program ImageJ. Oxyblotting has been described before and was performed with the OxyBlot Protein Oxidation Detection Kit (Millipore, S7150) following the manufacturer'sinstructions.Citation32 The detection of oxidized proteins was achieved with the SuperSignal® West Femto Maximum Sensivity Substrate (Thermo, 34095). For normalization, an actin western blot respective whole protein staining with Coomassie Blue was used. Mitochondria were isolated with the Qproteome Mitochondria Isolation Kit (Qiagen, 37612) according to the manufacturer'sinstructions.
Confocal laser scanning microscopy (CLSM).
For the determination of mitochondrial morphology, cells were stained with MTR (final concentration 25 nM) for 30 min. LAMP-1 and LAMP-2 were detected by using an anti-LAMP-1 (BD Bioscience PharMingen, 555798) and anti-LAMP-2 (BD Bioscience PharMingen, 555803) antibody. All microscopy analyses were performed with a Leica TCS SP5 confocal laser scanning microscope at the appropriate spectral settings and equipped with the objectives HCX PL APO lambda blue 63.0x, 1.40 OIL UV and HCX PL APO 63.0x, 1.31 GLYC 37°C UV that were controlled by the LAS AF scan software (version 1.8.2) (Leica). All live-cell experiments were performed at 37°C and 5% CO2 in a humidified chamber. Pictures were visualized with IMARIS 6.0.0 (BITPLANE Scientific Solutions). The quantification of fluorescence intensity was performed with ImageJ as described before.Citation35
Electron microscopy.
Cells were fixed with phosphate-buffered 2.5% glutaraldehyde for 1 h and stained with 1% osmium tetroxide, dehydrated in a graded series of ethanol and embedded in Epon812 (Sigma, 45345). Electron micrographs of thin sections stained with phospho-tungstic acid and uranyl-acetate were taken with a Hitachi H500 transmission electron microscope (Hitachi).
PEG fusion assay.
Stably transfected mtCFP HeLa were stained with MTR and irradiated. Directly afterwards they were co-seeded with stably transfected mtGFP HeLa and 4 h later fused with mtCFP-HeLa by replacing the medium drop-wise with fusion medium [40% (v/v) PEG (MW 1450; Polysciences, 00679-250)] in culture medium] as described before.Citation6 Fused cells were washed twice (medium without FCS) and then incubated in culture medium.
Quantification of the lysosomal compartment.
Cells were stained with monoclonal LAMP-2 antibody, and pictures were taken by CLSM. The area but not the fluorescence intensity of all lysosomes in one cell (the lysosomal area) and the total cell area were encircled (freehand tool) and quantified via ImageJ. The lysosomal area was normalized to the total cell area, resulting in the relative lysosomal area.
Semiquantitative RT-PCR.
RNA isolation and semiquantitative RT-PCR for fission and fusion factors has been described before.Citation8 For normalization, β-actin mRNA was used. Primers had the following sequences: PGC1α: sense: 5′-CCACAGATT-CAGACCAGTGCTACC-3′; anti-sense: 5′-CTGCCTGGA-GACCTTGATCTTGAC-3′; ATG5: sense: 5′-ATGACTAGC-CGGGGGAACACC-3′; anti-sense: 5′-CCAGTTTACCAT-CACTGCC-3′; ATG6 (Beclin1): sense: 5′-GAACCTCAGCCG-AAGACTGAA-GGTCACTG-3′; anti-sense: 5′-CCACTGTGC-CAGATGTGGAAGGTTGC-3′; ATG7: sense: 5′-GTTCAGTG-CTTTTGACATGAGTGCTCCCAC-3′; anti-sense: 5′-CTTCGTCCTTTGACCTTGGAAGAAATCACT-3′; ATG8F (LC3 B): sense: 5′-GAAGATCTCCGTCGGAGAAGACCTTC-3′; anti-sense: 5′-GCGAATTCTT-ACACTGACAATT-TCATCCC-3′; ATG10: sense: 5′-ATGGAAGAAGATGAG-TTCATTGGAGAAAAA-3′; anti-sense: 5′-AAGTACAAAAAA-GGGTTGCCCAAGTATTGG-3′; ATG12: sense: 5′-ATGACTAGCC-GGGACACC-3′; anti-sense: 5′-CCAGTTTAC-CATCACTGCC-3′.
Quantification of mtDNA integrity.
Total DNA was isolated from a total of 1 × 106 cells with the Flexi Gene DNA Kit (Qiagen, 51204) as described previously.Citation18 A 0.22-kb and an 8.9-kb fragment of mtDNA were amplified by PCR and after gel electrophoresis the relative amplification of the 8.9-kb fragment normalized to the 0.22-kb fragment was determined with the Easywin 32 software (Herolab) as parameter for mtDNA damage.Citation57
Quantification of the ROS content (DHE staining).
ROS content was analyzed by staining cells with dihydroethidium (DHE; Molecular Probes, D23107, final concentration 5 µM) for 30 min. To evaluate the ethidium fluorescence, pictures of fluorescent samples were taken with constant microscope settings and fluorescence intensity of the micrographs was quantified using ImageJ. Because cells were not homogenously distributed on the cover slide fluorescence intensity was related to the area covered by cells (“relative fluorescence intensity”). For each micrograph, each gray value was multiplied with the respective number of pixels. The total sum of these products was divided by the total sum of pixels.
Quantification of mitochondrial membrane potential (TMRE staining).
For determination of the mitochondrial membrane potential, cells were stained with TMRE (Invitrogen, T-669, final concentration 100 nM) for 10 min and analyzed by CLSM. The quantification of the membrane potential was performed with the program ImageJ as described previously.Citation19 For each condition 20 pictures with at least three cells/picture were analyzed.
ATP measurement.
The ATP content was determined in triplicate by using the ViaLight Plus Kit (Lonza, LT07-321) according to the manufacturer's instructions. ATP values were normalized to the whole protein amount after gel electrophoresis and Coomassie Blue staining of the corresponding samples.
Quantification of lysosomal activity.
The lysosomal activity was quantified by determining the activity of acid phosphatase, a lysosomal key enzyme. Experiments were performed in triplicate in a 96-well plate. 200,000 cells were lysed in citrate buffer and added to the substrate provided by the Acid Phosphatase Assay Kit (Sigma, CS0740). After 30 min incubation at 37°C the absorbance of the p-nitrophenol was determined at 405 nm in an ELISA reader (Tecan). The OD of non-transfected cells was set as 100%.
Statistics.
Results are expressed as means ± s.e.m. of n experiments, apart from the growth curves where SD was used. ANOVA was used to compare sets of data. Differences were considered statistically significant when p < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| ATG5 | = | autophagy-related protein 5 |
| ATG12 | = | autophagy-related protein 12 |
| CEF | = | chicken embryonic fibroblasts |
| CLSM | = | confocal laser scanning microscopy/microscope |
| HUVEC | = | human umbilical vein endothelial cells |
| LAMP | = | lysosomal-associated membrane protein |
| LC3B | = | microtubule-associated protein 1 light chain 3 beta (ATG8F) |
| MTR | = | MitoTracker Red CMX ROS |
| PD | = | population doublings |
| QC | = | quality control |
| RCAS | = | retroviral replication-competent avian leukosis virus long terminal repeat, splice acceptor |
| ROS | = | reactive oxygen species |
Figures and Tables
Figure 1 Mitochondrial dynamics does not act in early quality control after ROS-induced mitochondrial damage. (A) Young HUVEC were treated for 10 min with hydrogen peroxide (final concentration 13.2 mM) or were stained with MTR and irradiated for 15 or 30 min. Immediately after treatment the amount of oxidized proteins in mitochondrial and whole cell lysates was analyzed by oxyblotting. After irradiation, mitochondrial lysates contained a significantly higher amount of oxidized proteins compared with whole cell lysates. In contrast, hydrogen peroxide-treated cells showed a much higher content of oxidized proteins in the whole cell lysate compared with the mitochondrial fraction; n = 3; p < 0.05. (B) Mitochondria in HeLa cells transfected with mtCFP were stained with MTR and either irradiated or not treated, subsequently co-cultured with nonirradiated mtGFP transfected HeLa cells and after 4 h fused by PEG. Cycloheximide (CHX) was added 30 min before PEG fusion to prevent the influence of ongoing protein synthesis. Samples were fixed at the indicated time points after PEG treatment and depicted by CLSM. While in controls complete mixing of the fluorescent proteins was observed, no fusion of fragmented mitochondria in irradiated cells took place. Color coding: green, GFP; blue, CFP; red, MTR; magenta, GFP, CFP and MTR; white, MTR plus CFP; yellow, MTR and GFP. (C) The PEG fusion experiments were quantified by Pearson correlation. A Pearson correlation factor of 1 means 100% colocalization, while low and negative values stand for lack of colocalization and strict separation, respectively. In control cells, an almost complete colocalization of mtCEF/MTR and mtGFP was observed while no colocalization of mtCEF/MTR and mtGFP was detected in the irradiated samples; n = 5−11; p < 0.05.
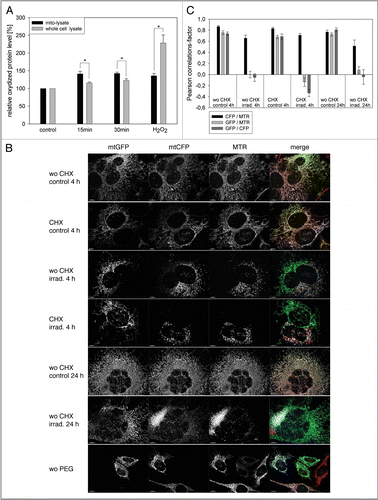
Figure 2 Mitophagy is the primary QC mechanism after ROS-induced mitochondrial damage. (A) HUVEC were transiently transfected with GFP-Parkin, irradiated after staining with MTR, and analyzed by CLSM at the indicated time points. In nontreated (nt) cells GFP-Parkin never associated with mitochondria at any time. In contrast, GFP-Parkin translocated in irradiated cells 2 h after irradiation to fragmented mitochondria, while 8 h after irradiation GFP-Parkin was again almost completely situated in the cytosol. (B) LC3-II, LC3-I and actin protein levels were determined in irradiated and control cells by western blotting at the indicated time points. (C) LC3-II western blots as shown representatively in (B) were normalized to actin protein content. The LC3-II/actin ratio of nonirradiated control cells was set as 1 for each time point and the LC3-II/actin ratio of irradiated cells was related to the respective control at the same time point. A time-dependent increase of the LC3-II/actin ratio after irradiation compared with nonirradiated control cells occurred; n = 6; p < 0.05. (D and E) Starting from 24 h after irradiation MTR-stained mitochondrial clusters appeared that could not be re-stained with MitoTracker Green, indicating loss of membrane potential (D, representative picture taken 72 h post irradiation). Quantification of the area of mitochondrial clusters demonstrated their increase until 72 h after irradiation (E); at least 100 cells/sample; n = 9−52; p < 0.05. (F and G) The lysosomal area was visualized by LAMP-2 staining in nonirradiated control cells and irradiated cells as shown in the representative pictures in (F) and related to the cell area (G). The relative lysosomal area of nonirradiated cells was set to 100%. An increase of the lysosomal area 72 h after irradiation compared with nonirradiated cells and irradiated cells at 0 h is discernable; n = 10−22; p < 0.05. (H and I) The amount of LAMP-2 protein was determined at the indicated time points by western blotting as shown by a representative blot (H) and normalized to actin protein levels. The quantification of the lysosomal protein showed a significant phasal upregulation after irradiation compared with nonirradiated controls (I); n = 3−4; p < 0.05.
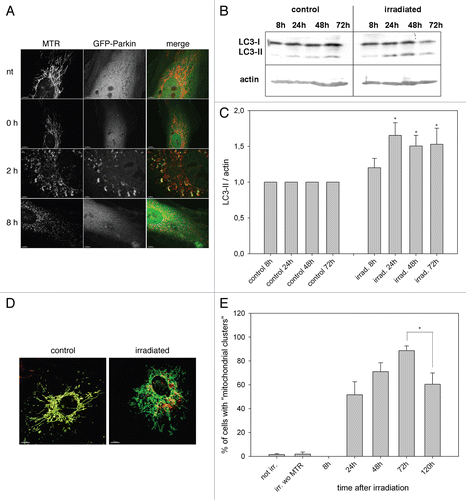
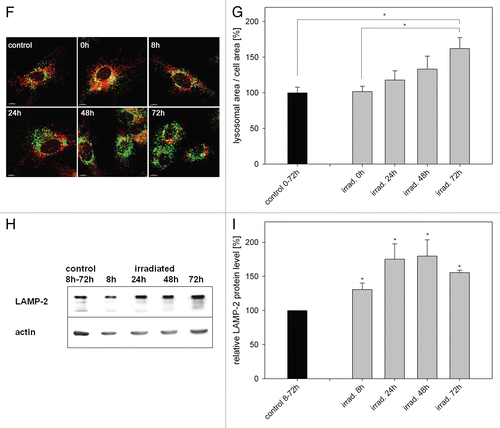
Figure 3 Overexpression of ATG5, LC3B and ATG12 increased life span in two cell models. (A) Young HUVEC were stained with MTR and irradiated (irrad) or nonirradiated (control). At the indicated time points after irradiation, mRNA levels were quantified by semiquantitative RT-PCR and normalized to the nonirradiated control. PGC1α, ATG5, ATG12 and LC3B were upregulated 8 h and 72 h after irradiation; n = 3−6; p < 0.05. (B) MTR-stained HeLa cells were transfected with plasmids expressing GFP-LC3B, GFP-ATG5 and GFP as control. 48 h after transfection the MTR fluorescence was quantified. The MTR fluorescence of GFP-LC3B and GFP-ATG5 was significantly decreased compared with GFP-expressing cells; n = 3; p < 0.05. (C) HUVEC stably transfected with GFP, GFP-ATG5, GFP-LC3B, GFP-ATG12 and LAMP-1-GFP were stained with MTR and analyzed by CLSM. In LC3B-, ATG5-and to a lesser amount ATG12-overexpressing senescent cells, a cytosolic GFP fluorescence with fluorescent autophagosomes was discernible; in LAMP-1 transfected cells the lysosomes are clearly labeled. (D) Young HUVEC (PD13) that were either nontransfected or stably transfected with GFP, GFP-ATG5, GFP-LC3B GFP-ATG12 and LAMP-1-GFP were cultivated in triplicate until they reached replicative senescence. All autophagy protein-expressing cells exhibited a significantly extended life span and higher PD numbers compared with control cells (non-transfected and GFP-overexpressing cells). In contrast LAMP-1-overexpressing HUVEC reached senescence after a similar cultivation time as the control cells, indicating that enhanced expression of autophagy proteins prolonged the replicative life span of HUVEC; p < 0.05. (E) Young CEF in passage 8 were infected with virus coding either for GFP or GFP-gLC3B (see inset) and cultivated in triplicate until they reached replicative senescence. Overexpression of GFP-gLC3B resulted in a prolonged replicative life span and significantly increased PD number in comparison to GFP-transfected cells, in accordance with the results obtained for HUVEC presented in ;p < 0.005. (F) Pre-senescent HUVEC (PD 22) were either nontransfected or stably transfected with plasmids expressing GFP, GFP-ATG5, GFP-LC3B GFP-ATG12 or LAMP-1-GFP. Cells were cultivated in triplicate until they reached replicative senescence. In contrast to young HUVEC (), no expansion of the life span or the PD number became apparent, indicating the absence of a rejuvenating effect.
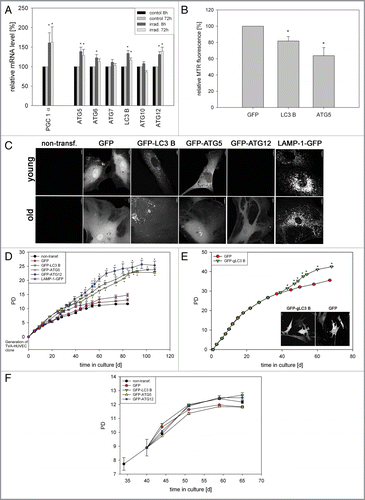
Figure 4 Overexpression of LC3B, ATG5, ATG12 and LAMP-1 mediates cell protective effects. (A and B) Young HUVEC expressing the indicated GFP fusion proteins (A) or young CEF overexpressing GFP and GFP-gLC3B (B) were treated with increasing doses of hydrogen peroxide for 10 min and the relative cell number of surviving adherent cells was determined and normalized to the untreated cells 24 h after treatment. Overexpression of the autophagy genes or LAMP-1 protected against hydrogen peroxide-induced cell death; (A) n = 4−6; p < 0.05; * = significant to GFP transfected cells, ** = significant to nontransfected cells; (B) n = 8; p < 0.05. (C and D) Young HUVEC stably overexpressing the indicated proteins (C) and young CEF overexpressing GFP and GFP-gLC3B (D) were stained with the mitochondrial membrane potential indicator TMRE and depicted by microscopy with constant settings. The TMRE fluorescence intensity was normalizedtothe fluorescence intensity of nontransfected HUVEC, and GFP transfected CEF, respectively. Overexpression of the autophagy genes and to a lesser amount LAMP-1 enhanced the mitochondrial membrane potential in both cell models; (C) n = 4−9; p < 0.05, (D) n = 7; p < 0.05. (E) Young HUVEC were stably transfected with GFP, GFP-ATG5, GFP-ATG12, GFP-LC3B and LAMP-1-GFP. Their ATP content was normalized to the GFP-transfected cells. Overexpression of the autophagy genes but not of LAMP-1 enhanced the ATP content; n = 3−4; p < 0.05. (F) A large fragment of the mtDNA (8.9 kb) was amplified by PCR and normalized on an amplified small fragment of the mtDNA (0.2 kb). The relative amplification of the 8.9-kb fragment acts as parameter for mtDNA integrity. The quantification in nontransfected and GFP-LC3B-overexpressing HUVEC at different time points of their growth curve shows an increased relative amplification of the 8.9-kb fragment after LC3B overexpression, indicating enhanced protection against mtDNA damage; 10–15 d (young cells): n = 3; 50–60 d (pre-senescent): n = 6; p < 0.05.
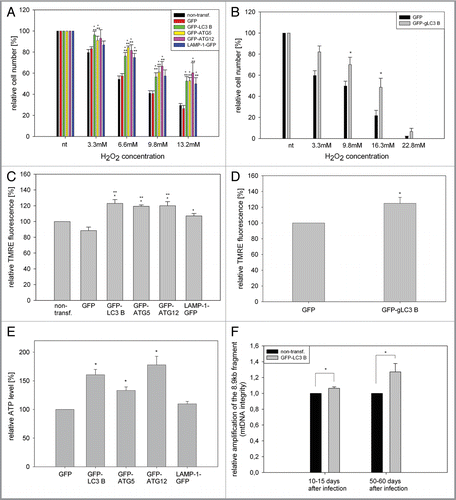
Figure 5 Overexpression of LC3B, ATG5 and ATG12 results in accumulation of oxidized material. (A and B) To determine the ROS content, young HUVEC stably overexpressing the indicated GFP-fusion proteins (A) or young CEF overexpressing GFP and GFP-gLC3B (B) were stained with DHE and the relative ethidium fluorescence was normalized to nontransfected HUVEC (A), or GFP transfected CEF (B). Overexpression of autophagy proteins or LAMP-1 did not alter the ROS levels in young HUVEC and CEF; n = 3−6. (C and D) The amount of oxidatively damaged proteins was analyzed by oxyblot in young HUVEC (C) and CEF (D) expressing the indicated GFP fusion proteins and normalized to actin (C) or total protein content (D). The amount of oxidized proteins of nontransfected cells or GFP-transfected (D) cells was set to 100%. A significant increase of carbonylated proteins was measured in cells overexpressing autophagy proteins in both cell types; (C) n = 4−7; p < 0.05, (D) n = 3; p < 0.05. (E and F) The amount of the active form of LC3, LC3-II, was determined by western blotting of nontransfected HUVEC and HUVEC overexpressing the indicated fusion proteins. A representative western blot is shown in (E). The quantification (F) shows after normalization to actin that overexpression of the autophagy proteins but not of LAMP-1 increases significantly the LC3-II/actin ratio compared with GFP and nontransfected cells; n = 6−8; p < 0.005. (G and H) Measurement of the lysosomal activity in nontransfected or overexpressing HUVEC (G), or CEF (H), respectively, was performed by quantifying the acid phosphatase activity. Only cells overexpressing LAMP-1-GFP demonstrated a significantly enhanced acid phosphatase activity; (G) n = 3−5; p < 0.05, (H) n = 5. (I) HUVEC were transiently transfected with either GFP and GFP-ATG5 or scrambled siRNA and a siRNA against ATG5. After 72 h the amount of oxidized proteins was determined by oxyblotting, normalized to actin protein expression and compared with the respective controls; transient overexpression of GFP-ATG5 increased significantly the amount of oxidized proteins compared with GFP-transfected cells; n = 3; p < 0.05.
Additional material
Download Zip (12.2 MB)Acknowledgments
We thank Monika Vöth for assistance with the electron microscopy. S.M. received a stipend from the CMP Frankfurt, which is thankfully acknowledged. We acknowledge grateful the support from the EU Integrated Project “MiMage” CT 2004-512020 and the BMBF project “GeronotoMitosys” (0315584A).
Related Research Data
References
- Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J 2008; 27:306 - 314; PMID: 18216873; http://dx.doi.org/10.1038/sj.emboj.7601972
- Bereiter-Hahn J, Jendrach M. Mitochondrial dynamics. Int Rev Cell Mol Biol 2010; 284:1 - 65; PMID: 20875628; http://dx.doi.org/10.1016/S1937-6448(10)84001-8
- Ono T, Isobe K, Nakada K, Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet 2001; 28:272 - 275; PMID: 11431699; http://dx.doi.org/10.1038/90116
- Nakada K, Inoue K, Ono T, Isobe K, Ogura A, Goto YI, et al. Intermitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med 2001; 7:934 - 940; PMID: 11479626; http://dx.doi.org/10.1038/90976
- Sato A, Nakada K, Shitara H, Yonekawa H, Hayashi J. In vivo interaction between mitochondria carrying mtDNAs from different mouse species. Genetics 2004; 167:1855 - 1861; PMID: 15342523; http://dx.doi.org/10.1534/genetics.103.021287
- Busch K, Bereiter-Hahn J, Wittig I, Schägger H, Jendrach M. Mitochondrial dynamics generate equal distribution but patchwork localisation of respiratory complex I. Mol Membr Biol 2006; 23:509 - 520; PMID: 17127623; http://dx.doi.org/10.1080/09687860600877292
- Malka F, Guillery O, Cifuentes-Diaz C, Guillou E, Belenguer P, Lombès A, et al. Separate fusion of outer and inner mitochondrial membranes. EMBO Rep 2005; 6:853 - 859; PMID: 16113651; http://dx.doi.org/10.1038/sj.embor.7400488
- Jendrach M, Mai S, Pohl S, Vöth M, Bereiter-Hahn J. Short-and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion 2008; 8:293 - 304; PMID: 18602028; http://dx.doi.org/10.1016/j.mito.2008.06.001
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008; 27:433 - 446; PMID: 18200046; http://dx.doi.org/10.1038/sj.emboj.7601963
- Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta 2008; 1777:860 - 866
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795 - 803; PMID: 19029340; http://dx.doi.org/10.1083/jcb.200809125
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010; 8:e1000298; PMID: 20126261; http://dx.doi.org/10.1371/journal.pbio.1000298
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007; 282:37298 - 37302; PMID: 17986448; http://dx.doi.org/10.1074/jbc.C700195200
- Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2004; 36:2503 - 2518; PMID: 15325588; http://dx.doi.org/10.1016/j.biocel.2004.05.009
- Eskelinen EL, Schmidt CK, Neu S, Willenborg M, Fuertes G, Salvador N, et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell 2004; 15:3132 - 3145; PMID: 15121881; http://dx.doi.org/10.1091/mbc.E04-02-0103
- Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J 2007; 26:313 - 324; PMID: 17245426; http://dx.doi.org/10.1038/sj.emboj.7601511
- Terman A, Gustafsson B, Brunk UT. Mitochondrial damage and intralysosomal degradation in cellular aging. Mol Aspects Med 2006; 27:471 - 482; PMID: 16973208; http://dx.doi.org/10.1016/j.mam.2006.08.006
- Jendrach M, Pohl S, Voth M, Kowald A, Hammerstein P, Bereiter-Hahn J. Morpho-dynamic changes of mitochondria during ageing of human endothelial cells. Mech Ageing Dev 2005; 126:813 - 821; PMID: 15888336; http://dx.doi.org/10.1016/j.mad.2005.03.002
- Mai S, Klinkenberg M, Auburger G, Bereiter-Hahn J, Jendrach M. Drp1 and Fis1 decrease mediates mitochondrial elongation in senescent cells and enhances resistance against oxidative stress via PINK1. J Cell Sci 2010; 124:917 - 927; http://dx.doi.org/10.1242/jcs.059246
- Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008; 4:176 - 184; PMID: 18059160
- Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal 2006; 8:152 - 162; PMID: 16487049; http://dx.doi.org/10.1089/ars.2006.8.152
- Lamming DW, Latorre-Esteves M, Medvedik O, Wong SN, Tsang FA, Wang C, et al. HST2 mediates SIR2-independent life-span extension by calorie restriction. Science 2005; 309:1861 - 1864; PMID: 16051752; http://dx.doi.org/10.1126/science.1113611
- Berdichevsky A, Viswanathan M, Horvitz HR, Guarente L. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell 2006; 125:1165 - 1177; PMID: 16777605; http://dx.doi.org/10.1016/j.cell.2006.04.036
- Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, et al. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 2008; 132:1025 - 1038; PMID: 18358814; http://dx.doi.org/10.1016/j.cell.2008.01.030
- Wang PY, Neretti N, Whitaker R, Hosier S, Chang C, Lu D, et al. Long-lived Indy and calorie restriction interact to extend life span. Proc Natl Acad Sci USA 2009; 106:9262 - 9267; PMID: 19470468; http://dx.doi.org/10.1073/pnas.0904115106
- Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 2005; 1:131 - 140; PMID: 16874025; http://dx.doi.org/10.4161/auto.1.3.2017
- Yen WL, Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda) 2008; 23:248 - 262; PMID: 18927201; http://dx.doi.org/10.1152/physiol.00013.2008
- Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, et al. The life span-prolonging effect of sirtuin-1 is mediated by autophagy. Autophagy 2010; 6:186 - 188; PMID: 20023410; http://dx.doi.org/10.4161/auto.6.1.10817
- Alvers AL, Wood MS, Hu D, Kaywell AC, Dunn WA, Aris JP. Autophagy is required for extension of yeast chronological life span by rapamycin. Autophagy 2009; 5:847 - 849; PMID: 19458476
- Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev 2009; 23:798 - 803; PMID: 19279323; http://dx.doi.org/10.1101/gad.519709
- Harman D. The biologic clock: the mitochondria?. J Am Geriatr Soc 1972; 20:145 - 147; PMID: 5016631
- Strecker V, Mai S, Muster B, Beneke S, Bürkle A, Bereiter-Hahn J, et al. Aging of different avian cultured cells is independent of ROS-induced damage. Mech Ageing Dev 2010; 131:48 - 59; PMID: 19948180; http://dx.doi.org/10.1016/j.mad.2009.11.005
- Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 2005; 280:20722 - 20729; PMID: 15778222; http://dx.doi.org/10.1074/jbc.M413934200
- Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA 2007; 104:14050 - 14055; PMID: 17709747; http://dx.doi.org/10.1073/pnas.0704014104
- Unterluggauer H, Hütter E, Voglauer R, Grillari J, Vöth M, Bereiter-Hahn J, et al. Identification of cultivation-independent markers of human endothelial cell senescence in vitro. Biogerontology 2007; 8:383 - 397; PMID: 17377850; http://dx.doi.org/10.1007/s10522-007-9082-x
- Van Laar VS, Arnold B, Cassady SJ, Chu CT, Burton EA, Berman SB. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet 2011; 20:927 - 940; PMID: 21147754; http://dx.doi.org/10.1093/hmg/ddq531
- Ali F, Ali NS, Bauer A, Boyle JJ, Hamdulay SS, Haskard DO, et al. PPARdelta and PGC1alpha act cooperatively to induce haem oxygenase-1 and enhance vascular endothelial cell resistance to stress. Cardiovasc Res 2010; 85:701 - 710; PMID: 19903700; http://dx.doi.org/10.1093/cvr/cvp365
- Chen SD, Lin TK, Yang DI, Lee SY, Shaw FZ, Liou CW, et al. Protective effects of peroxisome proliferator-activated receptors gamma coactivator1alpha against neuronal cell death in the hippocampal CA1 subfield after transient global ischemia. J Neurosci Res 2010; 88:605 - 613; PMID: 19774674
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999; 98:115 - 124; PMID: 10412986; http://dx.doi.org/10.1016/S0092-8674(00)80611-X
- Gutsaeva DR, Carraway MS, Suliman HB, Demchenko IT, Shitara H, Yonekawa H, et al. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci 2008; 28:2015 - 2024; PMID: 18305236; http://dx.doi.org/10.1523/JNEUROSCI.5654-07.2008
- Gamboa JL, Andrade FH. Mitochondrial content and distribution changes specific to mouse diaphragm after chronic normobaric hypoxia. Am J Physiol Regul Integr Comp Physiol 2010; 298:R575 - R583; PMID: 20007520; http://dx.doi.org/10.1152/ajpregu.00320.2009
- Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol 2007; 170:75 - 86; PMID: 17200184; http://dx.doi.org/10.2353/ajpath.2007.060524
- Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, et al. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010; 142:590 - 600; PMID: 20723759; http://dx.doi.org/10.1016/j.cell.2010.07.018
- Erusalimsky JD, Kurz DJ. Endothelial cell senescence. Handb Exp Pharmacol 2006; 176:213 - 248; PMID: 17001772; http://dx.doi.org/10.1007/3-540-36028-X_7
- Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, et al. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol 2010; 176:2092 - 2097; PMID: 20363920; http://dx.doi.org/10.2353/ajpath.2010.091050
- Andrejewski N, Punnonen EL, Guhde G, Tanaka Y, Lullmann-Rauch R, Hartmann D, et al. Normal lysosomal morphology and function in LAMP-1-deficient mice. J Biol Chem 1999; 274:12692 - 12701; PMID: 10212251; http://dx.doi.org/10.1074/jbc.274.18.12692
- Wagner M, Hampel B, Bernhard D, Hala M, Zwerschke W, Jansen-Durr P. Replicative senescence of human endothelial cells in vitro involves G1 arrest, polyploidization and senescence-associated apoptosis. Exp Gerontol 2001; 36:1327 - 1347; PMID: 11602208; http://dx.doi.org/10.1016/S0531-5565(01)00105-X
- Tiwari M, Bajpai VK, Sahasrabuddhe AA, Kumar A, Sinha RA, Behari S, et al. Inhibition of N-(4-hydroxyphenyl)retinamide-induced autophagy at a lower dose enhances cell death in malignant glioma cells. Carcinogenesis 2008; 29:600 - 609; PMID: 18174255; http://dx.doi.org/10.1093/carcin/bgm264
- Cheng Y, Qiu F, Guo ZM, Tashiro SI, Onodera S, Ikejima TY. Autophagy inhibits reactive oxygen species-mediated apoptosis via activating p38-nuclear factor-kappa B survival pathways in oridonin-treated murine fibrosarcoma L929 cells. FEBS J 2009; 276:1291 - 1306; PMID: 19187231; http://dx.doi.org/10.1111/j.17424658.2008.06864.x
- Bhutia SK, Dash R, Das SK, Azab B, Su ZZ, Lee SG, et al. Mechanism of autophagy to apoptosis switch triggered in prostate cancer cells by antitumor cytokine melanoma differentiation-associated gene 7/interleukin-24. Cancer Res 2010; 70:3667 - 3676; PMID: 20406981; http://dx.doi.org/10.1158/0008-5472.CAN-09-3647
- Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 2007; 129:983 - 997; PMID: 17540177; http://dx.doi.org/10.1016/j.cell.2007.03.045
- Dewaele M, Martinet W, Rubio N, Verfaillie T, de Witte PA, Piette J, et al. Autophagy pathways activated in response to PDT contribute to cell resistance against ROS damage. J Cell Mol Med 2011; 15:1402 - 1414; PMID: 20626525; http://dx.doi.org/10.1111/j.1582-4934.2010.01118.x
- Yang Y, Xing D, Zhou F, Chen Q. Mitochondrial autophagy protects against heat shock-induced apoptosis through reducing cytosolic cytochrome c release and downstream caspase-3 activation. Biochem Biophys Res Commun 2010; 395:190 - 195; PMID: 20361931; http://dx.doi.org/10.1016/j.bbrc.2010.03.155
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004; 429:417 - 423; PMID: 15164064; http://dx.doi.org/10.1038/nature02517
- Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci USA 2005; 102:17993 - 17998; PMID: 16332961; http://dx.doi.org/10.1073/pnas.0508886102
- Federspiel MJ, Hughes SH. Retroviral gene delivery. Methods Cell Biol 1997; 52:179 - 214; PMID: 9379950; http://dx.doi.org/10.1016/S0091-679X(08)60379-9
- Santos JH, Mandavilli BS, Van Houten B. Measuring oxidative mtDNA damage and repair using quantitative PCR. Methods Mol Biol 2002; 197:159 - 176; PMID: 12013794