Abstract
The molecular mechanisms by which arsenic (As3+) causes human cancers remain to be fully elucidated. Enhancer of zeste homolog 2 (EZH2) is the catalytic subunit of polycomb-repressive complexes 2 (PRC2) that promotes trimethylation of lysine 27 of histone H3, leading to altered expression of tumor suppressors or oncogenes. In the present study, we determined the effect of As3+ on EZH2 phosphorylation and the signaling pathways important for As3+-induced EZH2 phosphorylation in human bronchial epithelial cell line BEAS-2B. The involvement of kinases in As3+-induced EZH2 phosphorylation was validated by siRNA-based gene silencing. The data showed that As3+ can induce phosphorylation of EZH2 at serine 21 in human bronchial epithelial cells and that the phosphorylation of EZH2 requires an As3+-activated signaling cascade from JNK and STAT3 to Akt. Transfection of the cells with siRNA specific for JNK1 revealed that JNK silencing reduced serine727 phosphorylation of STAT3, Akt activation and EZH2 phosphorylation, suggesting that JNK is the upstream kinase involved in As3+-induced EZH2 phosphorylation. Because As3+ is capable of inducing miRNA-21 (miR-21), a STAT3-regulated miRNA that represses protein translation of PTEN or Spry2, we also tested the role of STAT3 and miR-21 in As3+-induced EZH2 phosphorylation. Ectopic overexpression of miR-21 promoted Akt activation and phosphorylation of EZH2, whereas inhibiting miR-21 by transfecting the cells with anti-miR-21 inhibited Akt activation and EZH2 phosphorylation. Taken together, these results demonstrate a contribution of the JNK, STAT3 and Akt signaling axis to As3+-induced EZH2 phosphorylation. Importantly, these findings may reveal new molecular mechanisms underlying As3+-induced carcinogenesis.
Introduction
Arsenic, a metalloid present in the Earth’s crust, exists in nature in three allotropic forms-α (yellow), β (black) and γ (gray), as well as in ionic forms.Citation1 Through natural and human activities, As3+, especially the trivalent form of As3+, (As3+), can be released into drinking water, soil and food, where it acts as one of the most important environmental pollutants. The level of arsenic exceeding 3,000–9,900 ppb (~40–130 μM) in ground water was observed frequently in some areas of West Bengal of India, Northern Vietnam, Thailand, Argentina, Mexico and the United States.Citation2,Citation3 Such a high concentration of As3+ that occurred either naturally or anthropogenically in ground water or drinking water is far beyond the dosage of As3+, usually from 1–20 μM, used in the majority of cellular or experimental animal studies. Currently, millions of people worldwide have been long-term exposed to environmental or occupational sources of As3+.Citation4 Chronic exposure to As3+ is known to cause many types of human diseases, such as cancers of the skin, bladder, lung and liver.Citation1 However, the exact mechanism underlying As3+-induced carcinogenesis remains to be fully elucidated. Several mechanisms have been proposed to be involved in As3+-induced carcinogenesis, including genetic changes, inhibition of DNA repair, induction of oxidative stress and aberrant activation of signal transduction pathways.Citation5 Mounting evidence has shown that As3+ can interfere with a series of signaling pathways, and this effect is partially mediated through the generation of reactive oxygen species (ROS). For example, As3+-mediated activation of the MAP kinases, including ERK, JNK and p38, has been observed to drive cell transformation, apoptosis and proliferation in multiple cell types.Citation6 Furthermore, the serine/threonine kinase Akt, a key oncogenic protein kinase important for cell transformation and neoangiogenesis, can be activated by As3+.Citation7,Citation8 Recent studies suggest that As3+-induced activation of Akt is mediated through the upregulation of miR-190, which, in turn, downregulates PHLPP, a negative regulator of Akt.Citation9 Furthermore, the upstream kinases, JNK and STAT3, may also contribute to Akt activation in cellular responses to As3+.Citation10
Enhancer of Zeste homolog 2 (EZH2) is the catalytic subunit of polycomb repressive complex 2 (PRC2), which mainly serves as a methyltransferase targeting lysine 27 of histone H3 (H3K27) for trimethylation, resulting in gene silencing.Citation11,Citation12 Studies of solid tumors revealed that EZH2 expression was commonly elevated in tumor cells, especially in prostate cancer, breast cancer and hepatocellular carcinoma,Citation13-Citation17 implicating important roles of EZH2 in tumorigenesis.Citation16,Citation18 Although the mechanism underlying the overexpression of EZH2 in various cancers remains unclear, studies have revealed that the overexpression of EZH2 is responsible for the repression of tumor suppressor genes, such as p16INK4aCitation19 and BRCA1.Citation20
Previous studies have shown that the function of EZH2 can be regulated by Akt-dependent phosphorylation. In breast cancer cells, Akt can directly interact with and phosphorylate EZH2 on serine 21 (S21) in response to insulin-like growth factor-1 (IGF-1).Citation21 The authors of this study suggested that the Akt-dependent phosphorylation of EZH2 impedes the methyltransferase activity of the PRC2 complex to methylate H3K27.Citation21 Akt has also been shown to phosphorylate EZH2 at S21 during the cellular response to estrogen receptor signaling.Citation22 Because we previously found that As3+ activates Akt and induces cell transformation,Citation7,Citation23 we hypothesized that Akt-mediated EZH2 S21 phosphorylation may be an important mechanism underlying As3+-induced carcinogenesis in humans. In the present study, we demonstrate that As3+ induces EZH2 S21 phosphorylation in human bronchial epithelial cells through JNK- and STAT3-dependent Akt activation. Considering that miR-21 is regulated by STAT3, our data further suggest that miR-21 may be involved in the signaling cascade leading to As3+-induced EZH2 S21 phoshporylation.
Results
As3+ induces phosphorylation of EZH2 at serine 21 (S21)
Previous studies have shown that EZH2 is phosphorylated at S21 by the activated Akt kinases in response to growth factors or estrogen.Citation21,Citation22 To determine whether As3+ is able to induce phosphorylation of EZH2 at S21, human bronchial epithelium-derived BEAS-2B cells were treated with different concentrations of As3+ for 4 h. We previously found that As3+ is capable of inducing the activation of both JNK and Akt in BEAS-2B cells.Citation7,Citation24 Accordingly, we also examined the activation status of JNK and Akt in response to As3+ treatment when we study EZH2 phosphorylation. As depicted in , As3+ induced dose-dependent EZH2 S21 phosphorylation (pS21). pS21 EZH2 was barely detectable under basal conditions; however, pS21 EZH2 was detected in the cells treated with 10 μM As3+ for 4 h and peaked in the cells treated with 80 μM As3+. In contrast, As3+ did not affect the level of unphosphorylated EZH2. As an enzymatic subunit of the PRC2, the key function of EZH2 is to induce trimethylation of lysine 27 at the N terminus of the histone H3 proteins (H3K27me3). To determine whether As3+-induced EZH2 S21 phosphorylation affects the methylation status of histone H3, we also measured the level of H3K27me3 in the cells treated with As3+. As indicated in , we failed to detect notable changes in H3K27me3 during the cellular response to As3+, possibly because only a small fraction of EZH2 was phosphorylated. In parallel, we noted a dose-dependent As3+-induced activation of JNK and Akt.
Figure 1. As3+ induces EZH2 S21 phosphorylation and activation of JNK and Akt. (A) As3+ induces EZH2 S21 phosphorylation and activation of JNK and Akt in a dose-dependent manner. BEAS-2B cells were treated with the indicated doses of As3+ for 4 h and analyzed by western blotting using the indicated antibodies. H3K27me3, trimethylation of lysine 27 on histone H3; pEZH2, S21-phosphorylated EZH2. (B) Time-course studies of As3+-induced EZH2 S21 phosphorylation, activation of JNK and Akt. BEAS-2B cells were treated with 20 μM As3+ for the indicated times. Total cellular proteins were prepared for western blotting at the end of each treatment. (C) As3+-induced EZH2 S21 phosphorylation requires Akt activation. BEAS-2B cells were transfected with a negative control, 50 nM control siRNA (siCon) or 50 nM Akt siRNA (siAkt) as described in the Materials and Methods. Forty-eight hours after transfection, the cells were treated with 20 μM As3+ for 2 h. At the end of the cell culture period, cell lysates were prepared for determination of EZH2 S21 phosphorylation (pEZH2) and Akt activation by western blotting. Data are representative of at least three independent experiments.
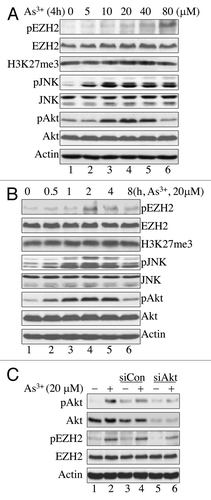
In a time-course experiment, EZH2 S21 phosphorylation peaked after 2 h of As3+ treatment (). The level of EZH2 S21 phosphorylation declined after the cells were treated with As3+ for 4–8 h. This finding correlates with the pattern of JNK and Akt activation induced by As3+ and indicates that JNK and Akt are possible upstream kinases involved in EZH2 S21 phosphorylation.
Earlier studies suggested that Akt can directly interact with and phosphorylate EZH2 at S21 in response to the growth factor IGF-1 or estrogen in breast cancer cells.Citation21,Citation22 Our data show that As3+ treatment induces concomitant activation of Akt and phosphorylation of EZH2 at S21; therefore, it is likely that As3+-induced EZH2 S21 phosphorylation requires Akt activation. To confirm that Akt is indeed responsible for As3+-induced EZH2 S21 phosphorylation, Akt was silenced by transfecting the cells with siRNA, specifically targeting Akt (siAkt). Cells transfected with control siRNA (siCon) were used as a negative control. As shown in , siAkt not only silenced total Akt protein and phospho-Akt, but also reduced the level of EZH2 S21 phosphorylation in the cells treated with As3+. This finding strongly supports the role of Akt in As3+-induced EZH2 S21 phosphorylation.
JNK inhibition diminishes As3+-induced EZH2 phosphorylation
Our initial experiments showed that As3+ treatment induced activation of JNK and Akt concomitantly with the EZH2 S21 phosphorylation. To determine whether JNK is the upstream kinase involved in As3+-induced EZH2 S21 phosphorylation, we pre-treated the cells with 20 μM JNK inhibitor SP600125 for 4 h, and then with 20 μM As3+ for 2 h. SP600125 is a relatively specific inhibitor that suppresses the phosphorylation and activation of JNK.Citation25 As indicated in , pre-treatment of the cells with SP600125 substantially reduced the As3+-induced activation of both JNK and Akt. This treatment also resulted in a significant decrease in As3+-induced EZH2 S21 phosphorylation, suggesting that JNK is indeed the upstream kinase responsible for As3+-induced EZH2 phosphorylation.
Figure 2. EZH2 phosphorylation at S21 is JNK-dependent. (A) JNK inhibition blocked As3+-induced Akt activation and EZH2 S21 phosphorylation. BEAS-2B cells were cultured in the presence or absence of 20 μM SP600125 (SP), a JNK inhibitor for 4 h and then with 20 μM As3+ for an additional 2 h. (B) siRNA-mediated silencing of JNK1 diminished As3+-induced EZH2 S21 phosphorylation and activation of STAT3 and Akt. BEAS-2B cells were transfected with a negative control, 50 nM control siRNA (siCon), 50 nM JNK1 siRNAb (siJNK1b) or 50 nM JNK1 siRNAc (siJNK1c). Cell lysates were prepared for western blotting after treatment with or without 20 μM As3+ for an additional 2 h. Data are representative of three or four independent experiments.
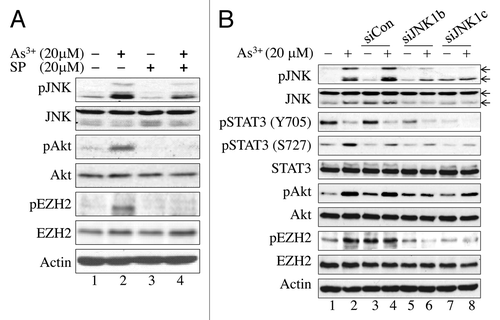
To further confirm the importance of JNK in As3+-induced EZH2 S21 phosphorylation, we next silenced JNK expression by transfecting the cells with two JNK1-specific siRNAs targeting different regions of the JNK1 mRNA. As expected, transfection of the cells with control siRNA did not affect As3+-induced activation of JNK and Akt or EZH2 S21 phosphorylation (lanes 3 and 4, ), whereas transfection of the cells with JNK1-specific siRNAs resulted in decreases in both As3+-induced activation of JNK and Akt and EZH2 S21 phosphorylation (lanes 5–8, ).
STAT3 mediates JNK-dependent Akt activation and EZH2 S21 phosphorylation
In response to growth signals or cellular stress, Akt is phosphorylated on threonine 308 (T308), which is located in the activation loop, and serine 473 (S473), which is located in the C-terminal hydrophobic motif.Citation26 Phosphorylation of S473 is a well-documented indicator of Akt activation in tumor cells or cancerous tissues. Because neither T308 nor S473 is the consensus phsophorylation site of the activated JNK kinase, it is likely that additional signaling molecules may be involved in the JNK-dependent Akt activation that leads to EZH2 S21 phosphorylation. Previous studies have suggested that STAT3 contributes to Akt activation upon ischemia/reperfusion injury or carcinogenic signaling.Citation27-Citation29 Additional evidence indicates that JNK can directly phosphorylate serine 727 (S727) of STAT3 to activate STAT3.Citation30 Accordingly, it is reasonable to hypothesize that STAT3 is the key mediator of As3+-induced JNK-dependent Akt activation and EZH2 S21 phosphorylation. To test this hypothesis, we examined As3+-induced STAT3 phosphorylation on tyrosine 705 (Y705) and S727 in cells transfected with JNK1-specific siRNAs (). As depicted in , As3+ induced STAT3 S727 phosphorylation. In contrast, STAT3 Y705 phosphorylation was inhibited by As3+ treatment (, lanes 2 and 4 in the panels of pY705, pS727 and STAT3). Although siRNA-mediated silencing of JNK1 has no effect on the total STAT3 protein levels, a notable reduction of As3+-induced STAT3 S727 phosphorylation was observed in cells in which JNK was partially silenced by JNK1-specific siRNAs (lanes 6 and 8, ). These data clearly indicate that As3+-induced, JNK-dependent Akt activation and EZH2 S21 phosphorylation are mediated by STAT3 S727 phosphorylation.
To confirm that STAT3 S727 phosphorylation is important for As3+-induced Akt activation and EZH2 S21 phosphorylation, we also pre-treated the cells with 20 μM STAT3 inhibitor V (Stattic, STAT3 In) for 4 h, and then with 20 μM As3+ for 2 h. Inhibition of STAT3 not only prevented As3+-induced STAT3 S727 phosphorylation, but also decreased As3+-induced Akt activation and EZH2 S21 phosphorylation (lane 4, ). This effect was not observed in the cells treated with DMSO as a vehicle control (lane 6, ).
Figure 3. Involvement of STAT3 in As3+-induced Akt activation and EZH2 S21 phosphorylation. (A) BEAS-2B cells were pretreated with a negative control, 20 μM STAT3 Inhibitor V (STAT3 In) or DMSO as a vehicle control for 4 h. The cells were then incubated in the presence or absence of 20 μM As3+ for an additional 2 h. (B) As3+ induces STAT3 reporter gene activity. BEAS-2B cells were transfected with a STAT3 reporter system containing a 40:1 mixture of a STAT3 firefly luciferase reporter vector and a renilla luciferase reporter vector. Twenty-four hours after transfection, the cells were treated with the indicated concentrations of As3+ for 6 h. Dual-luciferase activity was determined at the end of the cell culture period. The right panel shows a time-course study of As3+-induced STAT3 reporter gene activity. *: p < 0.05; **: p < 0.005. (C) As3+ reduces immunofluorescent signal of Y705-phosphorylated (pY705) STAT3. (D) As3+ induces nuclear accumulation of the S727-phosphorylated STAT3. Data are representative of three experiments.
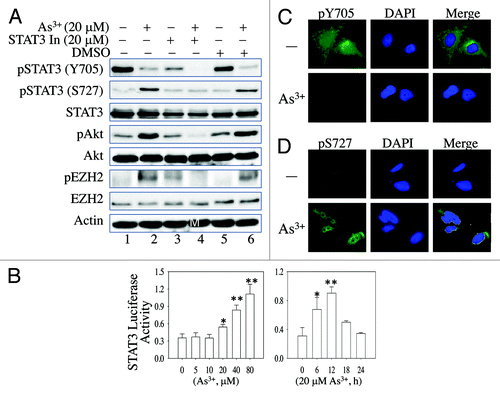
Previous studies provide evidence that STAT3 S727 phosphorylation is important for the nuclear translocation and transcriptional activity of the STAT3 protein, whereas Y705 phosphorylation might be involved in the dimerization of STAT3 proteins.Citation30 Considering that As3+ induces STAT3 S727 phosphorylation, it is possible that As3+ activates the transcriptional activity of STAT3. To determine whether As3+-induced S727 phosphorylation enhances the transcriptional activity of STAT3, we transfected the cells with a STAT3-based luciferase reporter system before treating them with As3+. Analysis of the reporter gene activity showed that As3+ stimulates STAT3 reporter gene activity in a dose-dependent manner (left panel, ), suggesting that As3+-induced S727 phosphorylation enhances the transcriptional activity of STAT3. Furthermore, in a time-course experiment in which the cells were treated with 20 μM As3+, STAT3 reporter activity was detected after 6 h of treatment with 20 μM As3+ and peaked after 12 h of treatment. The STAT3 reporter activity returned to basal levels after prolonged treatment of the cells with As3+ for 18–24 h (right panel, ). Further supporting this conclusion, immunofluorescent microscopy showed that As3+ decreased the overall Y705-phosphorylated STAT3 signal (), but increased the nuclear S727-phosphorylated STAT3 signal (). In contrast, As3+ treatment did not affect the expression level or the localization of unphosphorylated Stat3 (Fig. S1).
MiR-21 is involved in As3+-induced EZH2 phosphorylation through the regulation of Akt activation
STAT3 is considered an oncogenic transcription factor that regulates the expression of miRNAs and oncogenes important for cell growth or malignant transformation.Citation31 Previous studies in other experimental systems suggested that STAT3-regulated miRNA-21 (miR-21) is capable of inducing Akt activaion by downregulating protein translation of PTEN, an endogenous inhibitor of Akt.Citation29 To determine whether the expression of miR-21 was altered in the cells treated with As3+, total cellular RNAs were prepared for real-time PCR using probes specific for mature miR-21. As3+ treatment induced a 2.5-fold increase in miR-21 relative to the levels observed in the control cells (), suggesting that miR-21 may be involved in As3+-induced Akt activation and EZH2 S21 phosphorylation. This result also indirectly supported the observed increase in STAT3 transcriptional activity induced by As3+ ().
Figure 4. MiR-21 enhances As3+-induced Akt activation by downregulating Spry2. (A) As3+ induces miR-21 expression in BEAS-2B cells. The cells were cultured in the presence or absence of 20 μM As3+ for 6 h. The expression levels of miR-21 were determined by real-time PCR. (B) Overexpression of miR-21 precursor activates Akt and induces EZH2 S21 phosphorylation. Cells were transfected with 100 nM control miRNA or miR-21 precursor. EZH2 phosphorylation and Akt activation were determined by western blotting 24 h after transfection. (C) BEAS-2B cells were transfected with 30 nM anti-miR-21 (miR-21 inhibitor). Twenty-four hours later, the cells were incubated in the presence or absence of 20 μM As3+ for 2 h. EZH2 S21 phosphorylation and Akt activation were determined in cell lysates by western blotting. (D) Overexpression of miR-21 precursor downregulates Spry2. BEAS-2B cells were transfected with 100 nM control miR or 100 nM miR-21 precursor. The protein levels of PTEN, PCDC4 and Spry2 were determined 24 h after transfection. Data are representative of three experiments.
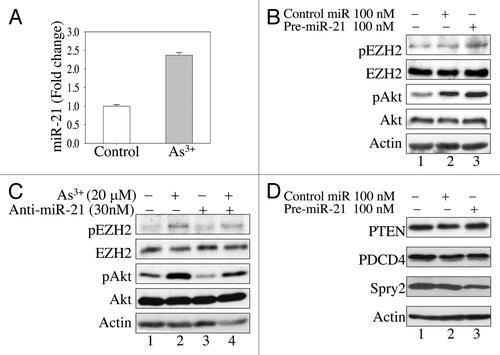
To provide direct evidence for the involvement of miR-21 in As3+-induced Akt activation and EZH2 S21 phosphorylation, we next transfected the cells with miR-21 precursor or an anti-miR that specifically inhibits the function of miR-21. An appreciable increase in Akt activation and EZH2 S21 phosphorylation was noted in the cells ectopically overexpressing miR-21 (lane 3, ). In contrast, transfection of the cells with an anti-miR that targets miR-21 partially abrogated As3+-induced activation of Akt and EZH2 S21 phosphorylation (lane 4, ). Taken together, these results suggest that STAT3 promotes As3+-induced Akt activation and EZH2 S21 phosphorylation through its downstream target, miR-21.
MiR-21 upregulates Akt and EZH2 S21 phosphorylation by downregulating Sprouty 2 (Spry2)
The key function of miRNAs is to fine-tune cellular signaling by suppressing protein translation or inducing the degradation of target mRNAs. As an oncogenic miRNA upregulated by STAT3, miR-21 has been implicated in the downregulation of tumor suppressors that antagonize Akt activation, such as PTEN and PDCD4,Citation29,Citation32 in some cells derived from tumor tissues. To determine whether PTEN or PDCD4 is also a target of miR-21 in epithelial cells, we first measured PTEN and PDCD4 protein levels in the cells transfected with miR-21 precursor. We failed to detect repression of PTEN or PDCD4 in the miR-21-overexpressing cells (lane 3, ), suggesting that the regulation of the target proteins by miR-21 might be cell context-dependent. Sprouty2 (Spry2), another tumor suppressor protein that regulates receptor tyrosine kinase (RTK) signaling, has been implicated as a possible target in miR-21-dependent Akt activation.Citation33,Citation34 In agreement with this notion, we observed a substantial downregulation of Spry2 in cells transfected with miR-21 precursor (lane 3, ). This result suggests that miR-21 enhances Akt activation in the As3+-treated cells by downregulating Spry2, another negative regulator of Akt signaling.
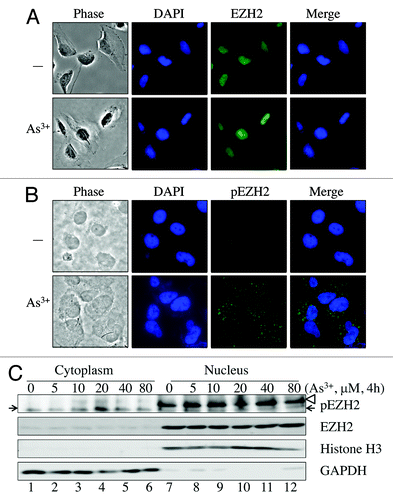
As3+ induces cytosolic localization of S21-phosphorylated EZH2
In breast cancer cells, Akt-dependent EZH2 S21 phosphorylation impairs the ability of EZH2 to methylate H3K27.Citation21 In bronchial epithelial cells, As3+ induces EZH2 S21 phosphorylation (). However, we did not observe a notable decrease in the levels of H3K27me3 in the cells treated with As3+, possibly because only a small fraction of EZH2 was phosphorylated (). Immunofluorescent staining revealed that unphosphorylated EZH2 is exclusively localized to the nucleus (), suggesting that EZH2 may display methyltransferase activity toward H3K27. Staining of the cells for S21-phosphorylated EZH2 showed that the basal level of S21-phosphorylated EZH2 is barely detectable. This finding supports the results of our western blotting data (). Immunofluorescent staining of S21-phosphorylated EZH2 revealed a striking punctuation or speckled pattern of the immunofluorescent signals for S21-phosphorylated EZH2 in the cells treated with As3+. These punctuated immunofluorescent signals for the S21-phosphorylated EZH2 were not uniform in size and were mostly distributed in the cytoplasm of the As3+-treated cells (). To verify the cytoplasmic location of the S21-phosphorylated EZH2 as determined by immunofluorescent staining, we have also performed cell fractionation studies. In agreement with the results obtained by immunofluorescent microscopy, the unphosphorylated EZH2 protein is exclusively presented in the nuclear fraction (lanes 7–12, ). For the S21-phosphorylated EZH2, we detected a dose-dependent increase of S21-phosphorylated EZH2 (pEZH2) in the cytoplasmic fraction (lanes 1–6, ), whereas nuclear pEZH2 was hardly detected. Considering the cytosolic localization of the S21-phosphorylated EZH2, it is likely that As3+-induced S21-phosphorylated EZH2 may have functions beyond its nuclear methyltransferase activity. EZH2 S21 phosphorylation may facilitate the dissociation of the PRC2 complex from chromatin and the translocation of the EZH2 protein from the nucleus to the cytoplasm, where it may interact with other cellular proteins.
Figure 5. As3+ induces cytosolic localization of S21-phosphorylated EZH2. (A) Immunofluorescent staining of EZH2 in control cells or cells treated with 20 μM As3+ for 2 h. (B) Immunofluorescent staining of S21-phosphorylated EZH2 (pEZH2) in cells cultured in the presence or absence of 20 μM As3+ for 2 h. Note the punctuation pattern of pEZH2 in the cytoplasm. (C) Nuclear or cytosolic localization of EZH2 and S21-phosphorylated EZH2 as determined by cellular fractionation. The purity of cytosolic fraction and nuclear fraction was validated by detecting GAPDH and Histone H3, respectively. The arrows indicate pEZH2; the blank triangle on the light of the top panel indicates non-specific bands in the nuclear fractions. Data are representative of three experiments.
Discussion
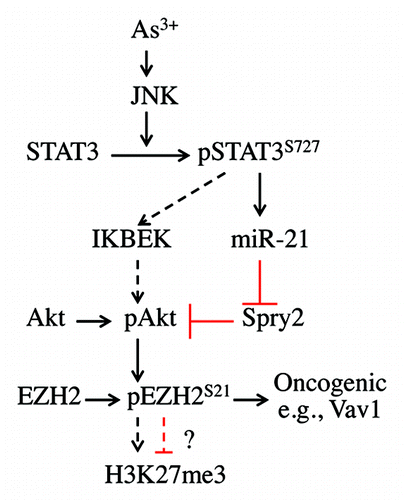
In the present study, we show that As3+ is capable of inducing Akt-dependent S21 phosphorylation of EZH2, the enzymatic subunit of the PRC2 complex responsible for the trimethylation H3K27me3. Moreover, siRNA-mediated silencing of Akt prevented As3+-induced EZH2 S21 phosphorylation. Interestingly, this As3+-induced response in epithelial cells resembles the responses induced by IGF-1 or estrogen in breast cancer cells.Citation21,Citation22 Furthermore, this work establishes a new signaling axis from JNK activation to STAT3 and miR-21 that results in the phosphorylation of EZH2 at S21 by Akt kinase (). Downregulation of JNK or STAT3 through the use of siRNAs or chemical inhibitors abrogated As3+-induced Akt activation and EZH2 S21 phosphorylation. An additional interesting finding from this study is the cytosolic localization of the S21-pohsphorylated EZH2 in As3+-treated cells, which possibly indicates that the methyltransferase activity of EZH2 is decreased after S21 phosphorylation.
Figure 6. Signaling cascade of As3+-induced EZH2 S21 phosphorylation. In response to As3+ treatment, activated JNK phosphorylates STAT3 at S727 to enhance the transcriptional activity of STAT3, leading to increased expression of miR-21, which, in turn, downregulates the expression of Spry2, a negative regulator of Akt signaling. The activated Akt phosphorylates EZH2 at S21, possibly causing the dissociation of PRC2 complex from chromatin and the cytosolic localization of S21-phosphorylated EZH2. JNK-activated STAT3 may also upregulate the expression of IKBKE, which can directly phosphorylate and activate Akt (dashed arrows), leading to EZH2 S21 phosphorylation. Following Akt-dependent phosphorylation on S21, the phosphorylated EZH2 may weaken H3K27me3 in nuclei or, alternatively, activate oncogenes, such as Vav1, as previously suggested, in the cytoplasm.
Accumulating evidence clearly indicates that environmental or occupational exposure to As3+ is a key risk factor for human cancers.Citation1 We and others have demonstrated that As3+ is a potent activator of signaling networks involving a number of intracellular kinases, cell cycle regulatory proteins and transcription factors that control cell fate, such as JNK,Citation35 GADD45α,Citation24 CDC25C,Citation36 Akt,Citation7,Citation8 NF-κB,Citation24 AP-1Citation37 and STAT3.Citation10 These networks are complex and interact both synergistically and antagonistically. For example, the activation of JNK is generally viewed as a stress response signal linked to cell death,Citation38 whereas STAT3, Akt, NFκB and AP-1 are mostly associated with cell growth or malignant transformation. However, depending on the cellular context, pathways with opposing functions might interact synergistically. It is also possible that certain signaling pathways display altered functions in the presence of simultaneous or asynchronous signals. Furthermore, some signaling pathways might be able to execute two contradictory functions in response to different environmental cues. The evidence supporting this notion is from the studies demonstrating bifurcate functions of the JNK pathways in cell apoptosis and compensatory proliferation.Citation39 Thus, it is likely that the growth-promoting role of the JNK pathways may be achieved through STAT3 S727 phosphorylation,Citation10 which, in turn, enhances Akt activation by upregulating miR-21, which represses negative regulators of Akt, such as PTEN, PDCD4 and Spry2 (). In addition to miR-21, STAT3 may elevate the expression of IκB kinase ε (IKBKE), which can directly phosphorylate Akt at both T308 and S473, leading to robust activation of the Akt kinases.Citation40-Citation42
Several lines of evidence suggest an association between aberrant expression of EZH2 and malignant disorders.Citation11 Overexpression of EZH2 is frequently observed in a wide variety of human cancers. EZH2 expression levels are strongly correlated with cancer cell proliferation, invasion and metastasis.Citation43 Considering its role as a methyltransferase that acts on H3K27, EZH2 has been viewed as a major repressor of the expression of tumor suppressors, including p16(INK4a), p19(ARF), Bim, CDKN1C (p57), E-cadherin and RUNX3.Citation11 EZH2 may be critical in maintaining the self-renewal capacity of normal or cancer stem cells by repressing lineage-specific differentiation programs.Citation44 In the past few years, several studies utilizing global proteomic profiling or biochemistry analyses have revealed that EZH2 is highly phosphorylated at specific serine or threonine residues. The first evidence of EZH2 phosphorylation was provided by Hung and colleagues,Citation21,Citation45 who demonstrated that Akt can directly phosphorylate EZH2 at S21 in response to IGF-1 in breast cancer cells. This phosphorylation of EZH2 at S21 was shown to decrease the affinity of PRC2 complex for chromatin and the methyltransferase activity of EZH2. Similarly, Akt-dependent EZH2 S21 phosphorylation was also observed in MCF-7 breast cancer cells in response to activation of the estrogen receptor (ER) by 17β-estradiol (E2) or the xenoestrogen diethylstilbestrol.Citation22 Consistent with the findings of Hung and colleagues, ER signaling-induced EZH S21 phosphorylation was shown to reduce the methyltransferase activity of EZH2 toward H3K27. In addition to S21, threonine 345 (T345, T350 in human EZH2) and 487 (T487, T492 in human EZH2) of EZH2 can be phosphorylated by cyclin-dependent kinase 1 (CDK1) and CDK2 during the mitotic phase of the cell cycle.Citation46-Citation49 It was believed that CDK1- or CDK2-mediated T345 phosphorylation enhances the recruitment of the PRC2 complex to specific loci, such as hox genes and the inactivation center on the X chromosomes, through binding of T345-phosphorylated EZH2 to ncRNAs, HOTAIR and Xist RepA.Citation47,Citation48 Inconsistencies remain regarding the functional consequences of EZH2 T487 phosphorylation. Hung and colleagues found that T487 phosphorylation disrupts the molecular association of EZH2 with two other PRC2 subunits, SUZ12 and EED, and, consequently, weakens the methyltransferase activity of the PRC2 complex.Citation49 This result differs from the findings of by Kaneko et al.,Citation47 who observed that methyltransferase activity was not affected by EZH2 T487 phosphorylation.
In the present report, we observed that the level of H3K27me3 was not altered following Akt-mediated EZH2 S21 phosphorylation in response to As3+-induced JNK and STAT3 activation, whereas other studies clearly indicated a reduction in H3K27me3 after EZH2 S21 phosphorylation.Citation21 There are several possible explanations for this discrepancy. First, only a small fraction of EZH2 was S21 phosphorylated in the cells treated with As3+. This small fraction may not be sufficient to affect the overall methyltransferase activity of the unphosphorylated EZH2. Second, whether EZH2 S21 phosphorylation reduces the enzymatic activity of EZH2 might be cell context-dependent. Different stimuli, such as As3+, growth factors or estrogen, might activate different signaling networks, which, in turn, determine the unique pattern of serine and threonine phosphorylation of EZH2. Lastly, this discrepancy might be a result of the different cell types used in these experiments. Our experiments were performed in immortalized but untransformed cells derived from human bronchial epithelial cells,Citation23 whereas the others used breast cancer cells. Despite these differences, the observed cytosolic localization of As3+-induced S21-phosphorylated EZH2 may provide indirect evidence supporting the notion that EZH2 S21 phosphorylation facilitates the dissociation of the PRC2 complex from chromatin and consequently reduces the methyltransferase activity of EZH2 toward H3K27. Alternatively, the S21 phosphorylated EZH2 may affect the assembly dynamics of other epigenetic regulatory complexes, such as those involved in methylation and demethylation on the lysine 4, lysine 9 or lysine 36 of the histone H3 proteins.
EZH2 is an important regulator of the epigenetic landscape of the genome, which controls the maintenance of stem cells, the development of cell lineages, cell proliferation and tumorigenesis.Citation11,Citation50 To date, information regarding the regulation of the expression and function of EZH2 by extracellular signals in different cellular and environmental settings remains limited. The discovery that As3+ induces a signaling cascade from JNK activation to S21 phosphorylation of EZH2 may provide some mechanistic insights into how environmental factors contribute to the epigenetic regulation that is critical for cell growth or malignant transformation. The key question that needs to be answered is the role of S21-phosphorylated EZH2 in As3+- and other carcinogen-induced carcinogenesis. A popular hypothesis regarding EZH2-mediated cancer development is that the methyltransferase activity of EZH2 catalyzes the trimethylation of H3K27 that inhibits expression of tumor suppressors.Citation11 The protein kinase Akt is well-established as an oncogenic kinase involved in cell transformation, cancer cell invasion, metastasis and angiogenesis in tumor tissues. If Akt-dependent EZH2 S21 phosphorylation decreases the methyltransferase activity of EZH2, Akt activation should be viewed as an important event in the antitumor response, because it can potentially elevate the expression of tumor suppressors. However, it may be difficult to detect an anti-tumor function of Akt because of its prominent role in other pro-oncogenic pathways, such as cell transformation and angiogenesis. Alternatively, although S21-phosphorylation reduces the methyltransferase activity of EZH2, the S21-phosphorylated EZH2 may serve as a new oncogenic molecule by interacting with other oncogenic signaling pathways. If this is the case, the oncogenic function of S21-phosphorylated EZH2 is expected to occur in the cytoplasm, as we demonstrated that the majority of S21-phosphorylated EZH2 is localized to the cytosolic compartment following As3+ treatment.
The capability of As3+ in inducing oxidative stress responses of the cells or tissues has long been recognized. It is believed that the reactive oxygen species (ROS) induced by As3+ play key roles in activating a number of intracellular signaling pathways.Citation1,Citation51 The involvement of ROS in As3+-induced activation of the JNK-STAT3-Akt signaling axis that linked to EZH2 phosphorylation has been confirmed in our laboratory, based on the fact that antioxidant N-acetyl-cysteine (NAC) prevented JNK activation and EZH2 S21 phosphorylation in the cells treated with either As3+ or H2O2 (Li et al., unpublished). It is unclear at the present how S21 phosphorylation changes the intracellular distribution of the EZH2 protein, which lacks a classic nuclear export sequence (NES). Two earlier studies demonstrated interaction of EZH2 and Vav1, an oncogenic protein with activity of guanine nucleotide exchanging, in the cytoplasm of the T-lymphocytes.Citation52,Citation53 It was unknown if such an interaction was regulated by the S21 phosphorylation of the EZH2 protein. One possibility is that S21-phosphorylated EZH2 may potentiate the oncogenic activity of the Vav1 protein to regulate actin polymerization, cell growth and malignant transformation. Additional studies are being undertaken to identify the cytosolic proteins that interact with S21-phosphorylated EZH2 in the cellular responses to As3+ and other environmental hazards.
Materials and Methods
Cell culture
The human bronchial epithelial cell line BEAS-2B was obtained from the American Type Culture Collection (ATCC). The cells were maintained in a humidified incubator in Dulbecco's modified Eagle's medium (DMEM, Invitrogen), supplemented with 5% fetal bovine serum (Invitrogen), 1% penicillin and 1% L-Glutamine (Sigma) at 37°C in the presence of 5% CO2. Cell viability was determined using a Bright Line Counting Chamber (Hausser Scientific).
siRNA transfection
The cells were seeded into 6-well plates and allowed to attach and grow to 70% confluency. Akt-specific siRNA and control siRNA were purchased from Cell Signaling. JNK1-specific siRNAs were synthesized by Eurofins. JNK2-specific siRNA was purchased from Santa Cruz Technology. BEAS-2B cells (1 × 105/ml in 6-well tissue culture plates) were reverse transfected with 50 nM siRNAs using Lipofectamine RNAiMAX reagent (Invitrogen) according to the protocol provided by the manufacturer. Forty-eight hours after transfection, the cells were treated with As3+ for an additional 2–24 h.
JNK and STAT3 inhibitor
The cells were seeded into 6-well plates. When the cells reached 80% confluency (72 h after plating), the cells were treated with the JNK inhibitor SP600125 (Calbiochem) or a STAT3 inhibitor V (Stattic). Four hours after inhibitor treatment, the cells were treated with As3+. The final concentration of each inhibitor was 20 μM.
MicroRNA precursor and inhibitor
The cells were reverse transfected with has-miR-21 precursor (Ambion) using siPORT NeoFX transfection reagent (Ambion) according to the protocol provided by the manufacturer. The cells were seeded in 6-well plates at a density of 1 × 105/ml. When the cells reached 80% confluency, the cells were transfected with has-miR-21 precursor and anti-miR-21 (Ambion) using Lipofectamine 2000 (Invitrogen) according to the protocol provided by the manufacturer. Twenty-four hours after transfection, the cells were treated with As3+ and analyzed by western blotting at the end of culture.
Luciferase reporter assay
The cells were seeded in 24-well plates at a density of 5 × 104/well. A STAT3 luciferase reporter system consisting of a 40:1 mixture of a STAT3 Firefly luciferase vector and a Renilla luciferase reporter vector was purchased from QIAGEN. The cells were transfected with the reporter constructs using Lipofectamine 2000 (Invitrogen) 48 h after plating according to the protocol provided by the manufacturer. Twenty-four hours after transfection, the cells were subjected to As3+ treatment for 6 or 24 h, and analyzed by luciferase assay using the Dual Luciferase Reporter Assay System (Promega) and a Glomax 20/20 luminometer (Promega). Firefly luciferase activity was normalized by Renilla luciferase activity.
Cellular fractionation and western blotting
To determine subcellular distribution of EZH2 and phosphorylated EZH2 proteins in control and As3+-treated BEAS-2B cells, nuclear and cytosolic proteins were prepared using NE-PER Nuclear and Cytoplasmic Fractionation Kit purchased from Thermo Scientific. Fractions were then analyzed by western blotting. The purity of the nuclear and cytoplasmic fractions were validated by western blotting using anti-GAPDH antibody as cytoplasmic marker and anti-Histone H3 antibody as nuclear marker, respectively. For regular western blotting, total cellular proteins were prepared using RIPA cell lysis buffer (Cell Signaling). The lysates were then collected and subjected to ultrasonication and centrifugation. The supernatants were collected, and proteins were quantified using a SpectraMax spectrophotometer (MDA Analytical Technologies). LDS sample buffer (Invitrogen) and dithiothreitol were added to the supernatants before boiling of the samples. The samples were separated by 10% SDS-PAGE and transferred to PVDF membranes. The PVDF membranes were blocked with 5% nonfat dry milk in TBST and then incubated with primary antibodies at 4°C overnight. The membranes were washed with TBST (3 × 10 min), incubated in secondary antibodies at room temperature for 1 h and washed again with TBST (3 × 10 min). CDP-Star Reagent (New England Biolabs) was used to detect immunoblotting signals.
The primary antibodies used in these experiments include anti-phospho-JNK (Cell Signaling), anti-JNK (Cell Signaling), anti-phospho-STAT3 (Ser727) (Cell Signaling), anti-phospho-STAT3 (Tyr705) (Cell Signaling), anti-STAT3 (Cell Signaling), anti-phospho-Akt (Ser473) (Cell Signaling), anti-Akt (Cell Signaling), anti-phospho-EZH2 (Ser21) (Bethyl) and anti-EZH2 (Cell Signaling). AP-conjugated anti-rabbit IgG antibody (Cell Signaling) was used as the secondary antibody.
Immunofluorescent staining
BEAS-2B cells were seeded into 24-well plates at a density of 5 × 104 cells per well. After 24 h, the cells were treated with or without 20 μM As3+ for 2 h and then fixed with 4% formaldehyde in PBS for 15 min at room temperature. The cells were briefly washed with PBS, incubated in blocking buffer (1 × PBS containing 5% normal goat serum and 0.3% Triton X-100) for 1 h and then incubated with the indicated antibodies for 1 h. The cells were next incubated with Alexa Fluor 488- or FITC-conjugated goat anti-rabbit IgG (Invitrogen) for 1 h in the dark. One drop of Prolong Gold antifade reagent with DAPI (Invitrogen) was added to each well before image analysis.
Real-time PCR
miRNA was quantified by real-time PCR as reported previously.Citation9 Briefly, cellular RNA was isolated from untreated cells and cells treated with 20 μM As3+ for 6 h using a MirVana kit (Ambion). Expression of miR-21 was analyzed using TaqMan MicroRNA Assays from Applied Biosystems. Reverse transcription and real-time PCR were performed on total RNA isolates according to the manufacturer’s instructions. SnoRNA U47 was used as the endogenous control for calibration of miR-21.
Statistics
Either SigmaPlot 9.0 or Microsoft Excel were used for statistical analyses of the quantitative data. The data are expressed as the mean ± standard deviation (SD). In some experiments, Student’s t-tests were used to determine the statistical significance of differences between samples treated under different conditions. Differences were considered statistically significant when p < 0.05.
Additional material
Download Zip (907.4 KB)Acknowledgments
This research was supported by grants from the NIH R01 ES-017217, NIH R01 ES-020137 (F.C.) and start-up funds from Wayne State University (F.C.). J.L. was supported by the Chinese Scholarship Council and start-up funds from Wayne State University (F.C.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, et al. Arsenic: toxicity, oxidative stress and human disease. J Appl Toxicol 2011; 31:95 - 107; PMID: 21321970
- Bhattacharya P, Welch AH, Stollenwerk KG, McLaughlin MJ, Bundschuh J, Panaullah G. Arsenic in the environment: Biology and Chemistry. Sci Total Environ 2007; 379:109 - 20; http://dx.doi.org/10.1016/j.scitotenv.2007.02.037; PMID: 17434206
- Nordstrom DK. Public health. Worldwide occurrences of arsenic in ground water. Science 2002; 296:2143 - 5; http://dx.doi.org/10.1126/science.1072375; PMID: 12077387
- Gibb H, Haver C, Gaylor D, Ramasamy S, Lee JS, Lobdell D, et al. Utility of recent studies to assess the National Research Council 2001 estimates of cancer risk from ingested arsenic. Environ Health Perspect 2011; 119:284 - 90; http://dx.doi.org/10.1289/ehp.1002427; PMID: 21030336
- Tokar EJ, Benbrahim-Tallaa L, Ward JM, Lunn R, Sams RL 2nd, Waalkes MP. Cancer in experimental animals exposed to arsenic and arsenic compounds. Crit Rev Toxicol 2010; 40:912 - 27; http://dx.doi.org/10.3109/10408444.2010.506641; PMID: 20812815
- Qian Y, Castranova V, Shi X. New perspectives in arsenic-induced cell signal transduction. J Inorg Biochem 2003; 96:271 - 8; http://dx.doi.org/10.1016/S0162-0134(03)00235-6; PMID: 12888263
- Zhang Y, Bhatia D, Xia H, Castranova V, Shi X, Chen F. Nucleolin links to arsenic-induced stabilization of GADD45alpha mRNA. Nucleic Acids Res 2006; 34:485 - 95; http://dx.doi.org/10.1093/nar/gkj459; PMID: 16421274
- Wang Z, Yang J, Fisher T, Xiao H, Jiang Y, Yang C. Akt activation is responsible for enhanced migratory and invasive behavior of arsenic-transformed human bronchial epithelial cells. Environ Health Perspect 2012; 120:92 - 7; http://dx.doi.org/10.1289/ehp.1104061; PMID: 21954225
- Beezhold K, Liu J, Kan H, Meighan T, Castranova V, Shi X, et al. miR-190-mediated downregulation of PHLPP contributes to arsenic-induced Akt activation and carcinogenesis. Toxicol Sci 2011; 123:411 - 20; http://dx.doi.org/10.1093/toxsci/kfr188; PMID: 21750348
- Liu J, Chen B, Lu Y, Guan Y, Chen F. JNK-dependent Stat3 phosphorylation contributes to Akt activation in response to arsenic exposure. Toxicol Sci 2012; 129:363 - 71; http://dx.doi.org/10.1093/toxsci/kfs199; PMID: 22696236
- Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer 2012; 106:243 - 7; http://dx.doi.org/10.1038/bjc.2011.551; PMID: 22187039
- Islam AB, Richter WF, Lopez-Bigas N, Benevolenskaya EV. Selective targeting of histone methylation. Cell Cycle 2011; 10:413 - 24; http://dx.doi.org/10.4161/cc.10.3.14705; PMID: 21270517
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002; 419:624 - 9; http://dx.doi.org/10.1038/nature01075; PMID: 12374981
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 2003; 100:11606 - 11; http://dx.doi.org/10.1073/pnas.1933744100; PMID: 14500907
- Chang Q, Zhang Y, Beezhold KJ, Bhatia D, Zhao H, Chen J, et al. Sustained JNK1 activation is associated with altered histone H3 methylations in human liver cancer. J Hepatol 2009; 50:323 - 33; http://dx.doi.org/10.1016/j.jhep.2008.07.037; PMID: 19041150
- Asangani IA, Harms PW, Dodson L, Pandhi M, Kunju LP, Maher CA, et al. Genetic and epigenetic loss of microRNA-31 leads to feed-forward expression of EZH2 in melanoma. Oncotarget 2012; 3:1011 - 25; PMID: 22948084
- Koh CM, Iwata T, Zheng Q, Bethel C, Yegnasubramanian S, De Marzo AM. Myc enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and post-transcriptional mechanisms. Oncotarget 2011; 2:669 - 83; PMID: 21941025
- Hou P, Liu D, Dong J, Xing M. The BRAF(V600E) causes widespread alterations in gene methylation in the genome of melanoma cells. Cell Cycle 2012; 11:286 - 95; http://dx.doi.org/10.4161/cc.11.2.18707; PMID: 22189819
- Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev 2007; 21:49 - 54; http://dx.doi.org/10.1101/gad.1499407; PMID: 17210787
- Gonzalez ME, DuPrie ML, Krueger H, Merajver SD, Ventura AC, Toy KA, et al. Histone methyltransferase EZH2 induces Akt-dependent genomic instability and BRCA1 inhibition in breast cancer. Cancer Res 2011; 71:2360 - 70; http://dx.doi.org/10.1158/0008-5472.CAN-10-1933; PMID: 21406404
- Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005; 310:306 - 10; http://dx.doi.org/10.1126/science.1118947; PMID: 16224021
- Bredfeldt TG, Greathouse KL, Safe SH, Hung MC, Bedford MT, Walker CL. Xenoestrogen-induced regulation of EZH2 and histone methylation via estrogen receptor signaling to PI3K/AKT. Mol Endocrinol 2010; 24:993 - 1006; http://dx.doi.org/10.1210/me.2009-0438; PMID: 20351197
- Chang Q, Pan J, Wang X, Zhang Z, Chen F, Shi X. Reduced reactive oxygen species-generating capacity contributes to the enhanced cell growth of arsenic-transformed epithelial cells. Cancer Res 2010; 70:5127 - 35; http://dx.doi.org/10.1158/0008-5472.CAN-10-0007; PMID: 20516118
- Chen F, Lu Y, Zhang Z, Vallyathan V, Ding M, Castranova V, et al. Opposite effect of NF-kappa B and c-Jun N-terminal kinase on p53-independent GADD45 induction by arsenite. J Biol Chem 2001; 276:11414 - 9; http://dx.doi.org/10.1074/jbc.M011682200; PMID: 11150309
- Bogoyevitch MA, Arthur PG. Inhibitors of c-Jun N-terminal kinases: JuNK no more?. Biochim Biophys Acta 2008; 1784:76 - 93; http://dx.doi.org/10.1016/j.bbapap.2007.09.013; PMID: 17964301
- Fayard E, Xue G, Parcellier A, Bozulic L, Hemmings BA. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr Top Microbiol Immunol 2010; 346:31 - 56; http://dx.doi.org/10.1007/82_2010_58; PMID: 20517722
- Ke B, Shen XD, Ji H, Kamo N, Gao F, Freitas MC, et al. HO-1-STAT3 axis in mouse liver ischemia/reperfusion injury: regulation of TLR4 innate responses through PI3K/PTEN signaling. J Hepatol 2012; 56:359 - 66; http://dx.doi.org/10.1016/j.jhep.2011.05.023; PMID: 21756853
- Blando JM, Carbajal S, Abel E, Beltran L, Conti C, Fischer S, et al. Cooperation between Stat3 and Akt signaling leads to prostate tumor development in transgenic mice. Neoplasia 2011; 13:254 - 65; PMID: 21390188
- Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 2010; 39:493 - 506; http://dx.doi.org/10.1016/j.molcel.2010.07.023; PMID: 20797623
- Zhang Y, Liu G, Dong Z. MSK1 and JNKs mediate phosphorylation of STAT3 in UVA-irradiated mouse epidermal JB6 cells. J Biol Chem 2001; 276:42534 - 42; http://dx.doi.org/10.1074/jbc.M106044200; PMID: 11553624
- He G, Karin M. NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res 2011; 21:159 - 68; http://dx.doi.org/10.1038/cr.2010.183; PMID: 21187858
- Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008; 27:2128 - 36; http://dx.doi.org/10.1038/sj.onc.1210856; PMID: 17968323
- Sayed D, Rane S, Lypowy J, He M, Chen IY, Vashistha H, et al. MicroRNA-21 targets Sprouty2 and promotes cellular outgrowths. Mol Biol Cell 2008; 19:3272 - 82; http://dx.doi.org/10.1091/mbc.E08-02-0159; PMID: 18508928
- Lee SA, Ladu S, Evert M, Dombrowski F, De Murtas V, Chen X, et al. Synergistic role of Sprouty2 inactivation and c-Met up-regulation in mouse and human hepatocarcinogenesis. Hepatology 2010; 52:506 - 17; http://dx.doi.org/10.1002/hep.23681; PMID: 20683950
- Chen F, Castranova V, Li Z, Karin M, Shi X. Inhibitor of nuclear factor kappaB kinase deficiency enhances oxidative stress and prolongs c-Jun NH2-terminal kinase activation induced by arsenic. Cancer Res 2003; 63:7689 - 93; PMID: 14633691
- Chen F, Zhang Z, Bower J, Lu Y, Leonard SS, Ding M, et al. Arsenite-induced Cdc25C degradation is through the KEN-box and ubiquitin-proteasome pathway. Proc Natl Acad Sci USA 2002; 99:1990 - 5; http://dx.doi.org/10.1073/pnas.032428899; PMID: 11842186
- Zhang D, Li J, Gao J, Huang C. c-Jun/AP-1 pathway-mediated cyclin D1 expression participates in low dose arsenite-induced transformation in mouse epidermal JB6 Cl41 cells. Toxicol Appl Pharmacol 2009; 235:18 - 24; http://dx.doi.org/10.1016/j.taap.2008.11.002; PMID: 19059425
- Chen F. JNK-induced apoptosis, compensatory growth, and cancer stem cells. Cancer Res 2012; 72:379 - 86; http://dx.doi.org/10.1158/0008-5472.CAN-11-1982; PMID: 22253282
- Suissa Y, Ziv O, Dinur T, Arama E, Gerlitz O. The NAB-Brk signal bifurcates at JNK to independently induce apoptosis and compensatory proliferation. J Biol Chem 2011; 286:15556 - 64; http://dx.doi.org/10.1074/jbc.M110.193235; PMID: 21385866
- Xie X, Zhang D, Zhao B, Lu MK, You M, Condorelli G, et al. IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci USA 2011; 108:6474 - 9; http://dx.doi.org/10.1073/pnas.1016132108; PMID: 21464307
- Guo JP, Coppola D, Cheng JQ. IKBKE protein activates Akt independent of phosphatidylinositol 3-kinase/PDK1/mTORC2 and the pleckstrin homology domain to sustain malignant transformation. J Biol Chem 2011; 286:37389 - 98; http://dx.doi.org/10.1074/jbc.M111.287433; PMID: 21908616
- Guo J, Kim D, Gao J, Kurtyka C, Chen H, Yu C, et al. IKBKE is induced by STAT3 and tobacco carcinogen and determines chemosensitivity in non-small cell lung cancer. Oncogene 2012; In press http://dx.doi.org/10.1038/onc.2012.39; PMID: 22330135
- Crea F. Histone code, human growth and cancer. Oncotarget 2012; 3:1 - 2; PMID: 22287500
- Villasante A, Piazzolla D, Li H, Gomez-Lopez G, Djabali M, Serrano M. Epigenetic regulation of Nanog expression by Ezh2 in pluripotent stem cells. Cell Cycle 2011; 10:1488 - 98; http://dx.doi.org/10.4161/cc.10.9.15658; PMID: 21490431
- Zeng X, Chen S, Huang H. Phosphorylation of EZH2 by CDK1 and CDK2: a possible regulatory mechanism of transmission of the H3K27me3 epigenetic mark through cell divisions. Cell Cycle 2011; 10:579 - 83; http://dx.doi.org/10.4161/cc.10.4.14722; PMID: 21278485
- Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J Biol Chem 2011; 286:28511 - 9; http://dx.doi.org/10.1074/jbc.M111.240515; PMID: 21659531
- Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, et al. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev 2010; 24:2615 - 20; http://dx.doi.org/10.1101/gad.1983810; PMID: 21123648
- Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, et al. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat Cell Biol 2010; 12:1108 - 14; http://dx.doi.org/10.1038/ncb2116; PMID: 20935635
- Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol 2011; 13:87 - 94; http://dx.doi.org/10.1038/ncb2139; PMID: 21131960
- Marchesi I, Fiorentino FP, Rizzolio F, Giordano A, Bagella L. The ablation of EZH2 uncovers its crucial role in rhabdomyosarcoma formation. Cell Cycle 2012; 11:3828 - 36; http://dx.doi.org/10.4161/cc.22025; PMID: 22983009
- Chen F, Shi X. Intracellular signal transduction of cells in response to carcinogenic metals. Crit Rev Oncol Hematol 2002; 42:105 - 21; http://dx.doi.org/10.1016/S1040-8428(01)00211-6; PMID: 11923072
- Su IH, Dobenecker MW, Dickinson E, Oser M, Basavaraj A, Marqueron R, et al. Polycomb group protein ezh2 controls actin polymerization and cell signaling. Cell 2005; 121:425 - 36; http://dx.doi.org/10.1016/j.cell.2005.02.029; PMID: 15882624
- Hobert O, Jallal B, Ullrich A. Interaction of Vav with ENX-1, a putative transcriptional regulator of homeobox gene expression. Mol Cell Biol 1996; 16:3066 - 73; PMID: 8649418