Abstract
Cells are constantly challenged by DNA damage and protect their genome integrity by activation of an evolutionary conserved DNA damage response pathway (DDR). A central core of DDR is composed of a spatiotemporally ordered net of post-translational modifications, among which protein phosphorylation plays a major role. Activation of checkpoint kinases ATM/ATR and Chk1/2 leads to a temporal arrest in cell cycle progression (checkpoint) and allows time for DNA repair. Following DNA repair, cells re-enter the cell cycle by checkpoint recovery. Wip1 phosphatase (also called PPM1D) dephosphorylates multiple proteins involved in DDR and is essential for timely termination of the DDR. Here we have investigated how Wip1 is regulated in the context of the cell cycle. We found that Wip1 activity is downregulated by several mechanisms during mitosis. Wip1 protein abundance increases from G1 phase to G2 and declines in mitosis. Decreased abundance of Wip1 during mitosis is caused by proteasomal degradation. In addition, Wip1 is phosphorylated at multiple residues during mitosis, and this leads to inhibition of its enzymatic activity. Importantly, ectopic expression of Wip1 reduced γH2AX staining in mitotic cells and decreased the number of 53BP1 nuclear bodies in G1 cells. We propose that the combined decrease and inhibition of Wip1 in mitosis decreases the threshold necessary for DDR activation and enables cells to react adequately even to modest levels of DNA damage encountered during unperturbed mitotic progression.
Introduction
Following DNA damage, eukaryotic cells protect their genome integrity by activation of a conserved DNA damage response pathway (DDR) that temporally arrests progression through the cell cycle (checkpoint) and promotes DNA repair. Depending on the mode of DNA damage, DDR is triggered by activation of the phosphatidylinositol 3-kinase-related kinases, namely ATM and DNA-PK in case of double-stranded DNA breaks or ATR in case of diverse types of lesions, mainly associated with DNA replication.Citation1 Recent studies and phosphoproteomic screens identified several hundred ATM/ATR substrates, including also checkpoint kinases Chk1/2 and tumor-suppressor protein p53. Activation of checkpoint kinases Chk1/2 promotes a fast establishment of the checkpoint by a direct inactivation or degradation of cdc25-family phosphatases. Phosphorylation of p53 by ATM/ATR and Chk1/2 leads to its stabilization and promotes its binding to promoters resulting in extensive changes in gene expression. Checkpoint is further strengthened by a p53-dependent expression of the cdk inhibitor p21 and by transcriptional repression of mitotic inducers cyclin B and cdc25B. In parallel to the checkpoint arrest, chromatin flanking the DNA lesion is extensively post-translationally modified to allow recruitment of various proteins involved in DNA repair (recently reviewed in ref. Citation2). Rapidly after DNA damage, ATM/ATR kinases phosphorylate the histone variant H2AX at Ser139 (referred to as γH2AX), and this modification generates a docking site for a large adaptor protein MDC1.Citation3 In turn, various ubiquitin E3 ligase complexes are recruited to the site of damage and mediate ubiquitination of the chromatin. Recent studies identified multiple protein complexes that control DNA-damage induced histone ubiquitination, and these include E3 ligases RNF8,Citation4,Citation5 RNF168Citation6 and HERC2Citation7; E2-conjugating enzyme Ubc13 and E1 ubiquitin-activating enzyme UBA1.Citation8 Both histone phosphorylation and ubiquitination are necessary for recruitment of 53BP1 and BRCA1 proteins, essential regulators of non-homologous end joining and homologous recombination DNA repair pathways, respectively.Citation2,Citation9 Besides the well-established role in DNA repair of lesions caused by ionizing radiation (IR) or by genetic rearrangements during maturation of the immune system, for example, 53BP1 is also involved in protecting endogenous underreplicated DNA regions (such as fragile sites) during interphase.Citation10,Citation11 According to this model, progression through unperturbed mitosis may generate DNA damage in these underreplicated regions. Since ubiquitination does not occur on mitotic chromatin, 53BP1 cannot localize to sites of DNA damage during mitosis, while γH2AX remains and provides a marker of mitotic DNA damage.Citation12,Citation13 In the subsequent G1 phase, 53BP1 is recruited to the damaged regions and prevents undesired DNA end processing until lesion is repaired during the next S phase. However, the precise molecular mechanisms regulating formation of these endogenous 53BP1 nuclear bodies remain to be elucidated.
Various phosphatases have been implicated in timely inactivation of the DDR following DNA repair.Citation14 Among these, the PP2C family serine/threonine phosphatase Wip1 seems to play an essential role, since it can dephosphorylate multiple proteins involved in DDR signaling including, ATM, Chk1, Chk2, p53, mdm2 and γH2AXCitation15-Citation21 and, thus, is considered as a major regulator of the cellular DDR.Citation22 Since Wip1 is a direct transcriptional target of p53, its expression increases following genotoxic stress.Citation23 Early after DNA damage, translation of Wip1 mRNA is blocked by miR-16 preventing premature termination of the DDR.Citation24 Later during the DDR, when the bulk of the lesions had been repaired, increased abundance of Wip1 allows termination of the DDR and by a negative feedback loop inactivates p53. However, a basal Wip1 activity needs to be present throughout the G2 checkpoint to limit the p53-dependent transcriptional repression of cyclin B and to maintain the recovery competence.Citation25 Importantly, loss of Wip1 strongly protects knockout mice from cancer development, suggesting that deregulated Wip1 may act as an oncogene.Citation26-Citation28 In addition, amplification of the PPM1D gene (encoding Wip1) was identified in various human tumors, pointing toward a role of Wip1 in cancer development.Citation27,Citation29-Citation34 Whereas the role of Wip1 in termination of DDR is relatively well-known, molecular mechanisms that control its function are still poorly understood. Here, we investigated how Wip1 is regulated during the cell cycle and found that the level of Wip1 is low in G1, increases toward G2 and declines during mitosis. Besides regulation at the protein level, Wip1 is extensively post-translationally modified, which contributes to its inactivation during mitosis. Our findings offer an explanation for the observed activation of the DDR pathway during unperturbed mitosis without exposure to exogenous DNA damaging insults.Citation10
Results
Protein abundance of Wip1 peaks in G2 and declines during mitosis
To gain insight into the regulation of Wip1 protein levels during the cell cycle, we synchronized HeLa cells at G1/S transition by a double thymidine block and then released them to fresh media containing nocodazole to allow progression to and arrest in mitosis. We noticed that whereas Wip1 was detectable throughout the S and G2 phases, its expression dramatically declined at 10–12 h post-thymidine release when cells entered mitosis (). Interestingly, cells released into media without nocodazole progressed through mitosis to G1 phase after 12 h and expressed Wip1, suggesting that the observed decrease of Wip1 may reflect a regulatory mechanism specific to mitosis. The same staining pattern was observed using two antibodies recognizing distinct epitopes in Wip1, thus indicating that the low signal is unlikely to reflect masking of the epitopes in mitosis. In addition, similar behavior of Wip1 was observed in U2OS cells, suggesting that the low abundance of Wip1 in mitosis is not restricted to a particular cell type (data not shown). Since synchronization of cells with thymidine may cause undesired stress response and potentially impair protein expression, we aimed to develop a system that would allow investigation of asynchronously growing cells.Citation35 We made use of the published fluorescent, ubiquitination-based cell cycle indicator (FUCCI) and established a stable cell line expressing markers of G1 and S/G2 phases.Citation36 After fluorescence-activated sorting of asynchronously growing cells, we obtained fractions highly enriched in G1 and G2 cells (; Fig. S1). Notably, we observed that G2 cells expressed approximately 2-fold more Wip1 compared with G1 cells (). Since transcription of Wip1 is controlled by p38/MAPK-p53 and JNK/c-Jun stress-responsive pathways, we hypothesized that the moderate difference in expression of Wip1 in G1 and G2 phases may be masked in cells synchronized with thymidine.Citation23,Citation37
Figure 1. Wip1 protein abundance during the cell cycle. (A) HeLa cells were synchronized by a double thymidine block, released into fresh media supplemented or not with nocodazole, and samples were collected at 2-h intervals and probed with indicated antibodies. pSer10-H3 was used as a marker of mitotic entry; degradation of cyclin A as a marker of prometaphase and degradation of cyclin B as a marker of mitotic exit. (B) Asynchronously growing FUCCI indicator expressing U2OS cells were pretreated with Hoechst DNA dye and the following populations of cells were sorted: double-negative (DN) and single RFP-positive cells (RNF+); single GFP-positive cells (GFP+); double-positive (DP) cells and samples were analyzed by flow cytometry. Note that the DN/RFP+ population corresponds to cells with a low DNA content (G1 phase), whereas GFP+ population corresponds to 4 n cells (G2 phase) and DP show intermediate DNA content (S phase). (C) Populations of cells from (B) analyzed by immunoblotting. Cyclin D was used as a marker of G1, cyclin A and Plk1 as markers of G2. (D) U2OS and RPE cells were cotransfected by Wip1 shRNA plasmid (shWip1) together with a mCherry marker and probed with polyclonal Wip1 (sc20712) or monoclonal Wip1 (sc37625) antibodies and with DAPI. Neighboring untransfected cells were used as a control (mock). Shown is quantification of immunofluorescence staining in interphase and mitotic cells. Note higher Wip1 signal intensity in cells with higher DAPI corresponding to G2 cells and decreased Wip1 signal in mitotic cells. As additional control of signal specificity, cells were treated with Etoposide (2.5 µM for 16 h), which caused accumulation of G2 cells with high intensity of Wip1. (E) Asynchronously growing U2OS cells were fixed and probed with indicated antibodies and DAPI. Note decreased Wip1 signal in mitosis and absence of the signal on mitotic chromatin.
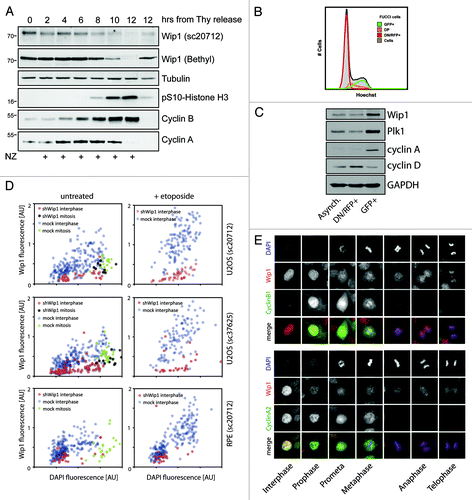
To substantiate our findings obtained by biochemical analysis of mixed cell populations, we set up an automated microscopic analysis of multiple individual cells. Total intensity of the DAPI signal was proportional to the DNA content and, as expected, was 2-fold higher in G2 cells compared with G1 cells. In addition, mitotic cells with condensed chromatin showed slightly higher DAPI signal compared with G2 cells. Remarkably, higher Wip1 staining intensity was found in interphase cells with higher DAPI signal compared with those with lower DAPI signal, thus supporting our conclusion that expression of Wip1 is higher in G2 cells compared with G1 cells (). Importantly, the observed immunofluorescence signal of Wip1 in interphase cells was specific, since it increased after treatment of cells with etoposide and was greatly reduced after depletion of Wip1 by RNAi (). Despite the commonly observed higher fluorescence staining background in mitosis, mitotic cells showed a lower intensity of the Wip1 staining compared with G2 cells, which is consistent with our analysis of the whole-cell lysates using immunoblotting ().
Recently, we reported that overexpressed EGFP-Wip1 is bound to chromatin throughout the cell cycle.Citation19 In good agreement with our previous findings, we observed that endogenous Wip1 was enriched in the nucleus during interphase (). In a striking contrast, we did not observe any specific signal of the endogenous Wip1 at the metaphase plate, suggesting that endogenous Wip1 might be absent from chromatin during mitosis (). To exclude the possibility that the antibody does not recognize the chromatin-bound Wip1 during mitosis, we examined cells expressing Wip1-mCherry and observed an identical staining pattern (Fig. S2).
Wip1 is degraded in mitosis by proteasome in APC/Ccdc20-dependent manner
Since our biochemical and microscopic analyses revealed decreased abundance of Wip1 in mitotic cells, we hypothesized that protein stability of Wip1 might be regulated during mitosis. To assess this possibility, we released cells from the thymidine block to a media supplemented with nocodazole and added a proteasomal inhibitor MG132 for the last 4 h. Interestingly, treatment with MG132 completely prevented the mitosis-associated decrease of Wip1, suggesting that Wip1 is normally degraded by the proteasome upon entry into mitosis (). Similarly, microscopic analysis of mitotic cells revealed that the intensity of the Wip1 staining increased after a short treatment with MG132 (). Finally, we wished to exclude the possibility that the low abundance of Wip1 in mitosis is caused by the absence of Wip1 mRNA. We synchronized cells in late G2 phase by treatment with a selective Cdk1-inhibitor Ro-3306 and released them into media with or without nocodazole for 3 h to obtain mitotic and G1 cells, respectively.Citation38 Similarly to the above experiments with the thymidine block/release synchronization, we noticed that protein levels of Wip1 declined in mitosis (). Importantly, both samples analyzed in parallel showed similar levels of Wip1 mRNA, suggesting that Wip1 abundance during mitosis is not regulated at the level of mRNA ().
Figure 2. Degradation of Wip1 during mitosis. (A) U2OS cells were synchronized by a double thymidine block, released into media with NZ and after 8 h, treated or not with 5 μM MG132. (B) Asynchronously growing cells were treated for 60 min with 5 µM RO-3306 to release cells from mitosis and prevent new cells from entering. Subsequently, cells were treated for 60 min with MG132 (5 μM) or DMSO, fixed, stained for Wip1 and the signal was quantified. Each dot corresponds to one cell. The dashed lines indicate the respective mean values. The difference between the means was analyzed by Student’s t-test (** p < 0.01). (C) Cells were synchronized in late G2 by Ro-3306 and released into fresh media with NZ for 2 h. Mitotic cells were collected by shake-off and analyzed by immunoblotting (left) or qPCR (right). (D) Cells were transfected with siRNAs targeting indicated proteins. One-third of the cells were synchronized for last 24 h with thymidine, and adherent cells were collected. One-third of the cells were treated with NZ for the last 12 h and collected by mitotic shake-off. Cell extracts were collected at 48 h after transfection and probed with indicated antibodies. RNA was extracted from the last part of cells and analyzed by qPCR (Fig. S3). (E) Cells were transfected with siRNAs targeting cdc20 or GAPDH (negative control). Mitotic cells were collected by shake-off 24 h after transfection and probed with indicated antibodies.
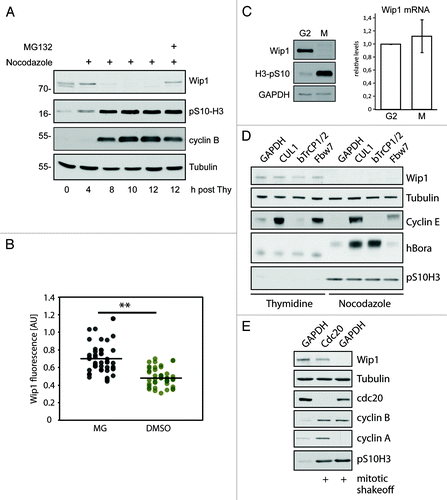
Next, we aimed to identify the mechanism of Wip1 degradation in mitosis. Given similar kinetics of cyclin A and Wip1 loss during prometaphase (), we argued that Wip1 might be degraded upon ubiquitylation by either SCF or APC/Ccdc20 E3 ubiquitin ligase complexes, both of which are active in early mitosis.Citation39 First, we depleted cells of an essential SCF complex component Cul1 and F-box proteins βTrCP1/2 or Fbw7 using siRNA and assayed the protein level of Wip1 in cells synchronized at G1/S transition or in mitosis (; Fig. S3). As expected, depletion of Cul1 or Fbw7 caused stabilization of the SCFFbw7 substrate cyclin E.Citation40 Similarly, depletion of Cul1 or βTrCP1/2 led to stabilization of hBora protein during mitosis.Citation41 Although we were able to efficiently deplete all indicated proteins, we did not observe any significant changes in levels of Wip1, either in interphase or in mitosis, suggesting that stability of Wip1 is not regulated by the SCF complex. Next, we depleted the APC/C activator cdc20 by RNAi and assayed the protein level of Wip1 in mitotic cells (; Fig. S3). As expected, efficient depletion of cdc20 enriched the population of mitotic cells and led to stabilization of cyclin ACitation42-Citation44 (). Importantly, depletion of cdc20 resulted in stabilization of Wip1 protein to levels comparable to those observed in interphase cells (). From these results we conclude that proteasomal degradation of Wip1 in mitosis is controlled by the APC/Ccdc20 complex.
Catalytic domain of Wip1 is phosphorylated at multiple sites in mitosis
Apart from the decreased protein level of endogenous Wip1, we noticed that an additional slower migrating band recognized by antibodies to Wip1 appeared in mitotic cell extracts, indicative of a possible post-translational modification. Indeed, Wip1-FLAG purified from mitotic cells showed a similar mobility shift that disappeared after treatment with lambda phosphatase, suggesting that Wip1 is phosphorylated during mitosis (). To better characterize the timing of the Wip1 phosphorylation during the cell cycle, we synchronized cells expressing a phosphatase-dead Wip1-D314A-FLAG fusion protein in G2 by adriamycine treatment and followed the occurrence of the slower migrating band corresponding to phosphorylated Wip1 during the progression to mitosis. Interestingly, behavior of Wip1 closely mimicked that of an established mitotic marker cdc27 (). Consistently, neither Wip1 nor cdc27 showed a mobility shift when mitotic entry was prevented by exposure of cells to the CDK inhibitor roscovitine, suggesting that phosphorylation of Wip1 occurs in mitosis (). In addition, the mobility shift of Wip1-D314A-FLAG rapidly disappeared when cells were released from nocodazole arrest and exited mitosis, further supporting the notion that such phosphorylation of Wip1 is restricted to mitosis (). To get insight into protein kinases responsible for modification of Wip1 during mitosis, we treated nocodazole-arrested cells expressing Wip1-FLAG with roscovitine or an MAPK inhibitor SB202190 and assayed the mobility of Wip1 separated on a PhosTag gel.Citation45 Whereas mitotic Wip1 migrates as three separate bands under these conditions, the band showing the slowest mobility disappeared upon treatment of cells with roscovitine (but not with the MAPK inhibitor), suggesting that Wip1 might be phosphorylated by Cdk1 during mitosis (). Consistent with this possibility, we observed no mobility shift in cells arrested in late G2 by exposure to the Cdk1 selective inhibitor Ro-3306 (data not shown). Moreover, bacterially expressed His-Wip1 was a good substrate for the Cdk1/cyclin B kinase in vitro ().
Figure 3. Wip1 is extensively phosphorylated in mitosis. (A) Expression of Wip1-FLAG was induced by tetracycline and Wip1-FLAG was immunoprecipitated from asynchronously growing cells (Asynch.), from mitotic cells arrested in NZ and collected by shake-off (Mitotic) or from cells treated with 0.5 μM adriamycine (Damage). Half of the immunopurified material was subjected to treatment with lambda phosphatase. (B) Cells expressing Wip1-D314A-FLAG were arrested in G2 by treatment with adriamycine and forced to enter mitosis by treatment with caffeine. Alternatively, roscovitine was added to prevent mitotic entry. (C) Cells expressing Wip1-D314A-FLAG were arrested in mitosis by treatment with NZ and released into fresh media for indicated time. Alternatively, mitotic exit was blocked by addition of MG132. (D) Cells expressing Wip1-FLAG were arrested in mitosis by NZ and treated with DMSO, roscovitine (12 μM) or p38i (3 μM, SB202190) for 90 min. Cell lysates were separated on Phostag SDS-PAGE and probed with indicated antibodies. (E) Phosphatase dead His-Wip1-D314A was subjected to in vitro kinase assay with Cdk1/cyclin B, separated on SDS-PAGE and phosphorylation was detected by autoradiography. (F) Wip1-D314A-FLAG was purified from mitotic cells and analyzed by MS. Scheme of identified phosphorylated residues is shown. (G) His-Wip1 or His-Wip1-S40A was subjected to in vitro kinase assay with Cdk1/cyclin B, separated on SDS-PAGE and phosphorylation was detected by autoradiography. (H) Cells expressing Wip1-FLAG, Wip1-S40A, Wip1-S46A or Wip1-AAA were synchronized in mitosis by NZ or grown asynchronously. Note a partial loss of mobility shift in the S40A mutant purified from mitotic cells. (I) Cells expressing Wip1-WT or Wip1–7A were grown asynchronously or arrested in mitosis by addition of nocodazole for 12 h and probed with indicated antibodies. Note decreased mobility shift of Wip1–7A in mitotic cells.
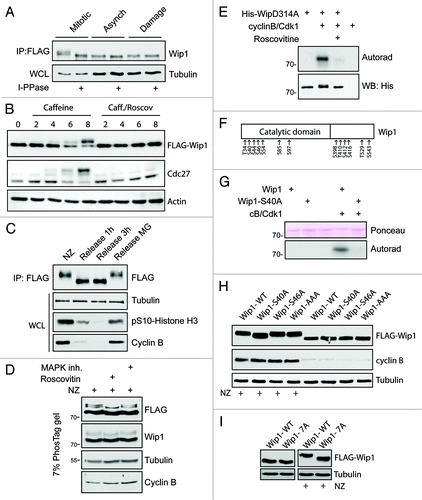
To identify the mitotically phosphorylated residues of Wip1, we purified a phosphatase-dead Wip1-D314A-FLAG from nocodazole-arrested cells and analyzed post-translational modifications by mass spectrometry. This analysis revealed seven phosphorylation sites within the catalytic domain of Wip1 and an additional six modified residues in the C-terminal domain (). Bioinformatic analysis of short linear motifs in the primary sequence of Wip1 revealed Ser40 as the only putative phosphorylation site by Cdk1.Citation46 Indeed, mutagenesis of Ser40 to alanine almost completely blocked the phosphorylation of Wip1 by Cdk1 in vitro, suggesting that Ser40 is the major phosphorylation site of Cdk1 (). In addition, Wip1-S40A mutant protein ectopically expressed in mitotic cells showed increased gel mobility compared with wild-type Wip1, confirming that Ser40 is phosphorylated also in vivo (). However, the observed partial impact of mutating Ser40 on the overall mobility shift in Wip1-S40A indicates that other phosphosites also exist on mitotic Wip1 (). Interestingly, mutagenesis of residues Thr410, Ser412 and Thr416 (identified as phosphosites by MS) to alanine did not affect the mobility of such Wip1-AAA protein in mitosis, suggesting that the extent of modification of these sites might be low, or such phosphorylations do not significantly affect Wip1 gel mobility (). In contrast, Wip1–7A mutant, in which all seven identified phosphorylated residues within the catalytic domain were mutated to alanine, showed a strongly reduced mobility shift in mitosis, suggesting that these residues are phosphorylated in mitosis (). Since Cdk1 phosphorylated only one of these seven phosphosites that we identified within the catalytic domain of Wip1, additional mitotic kinases likely also modify Wip1 during mitosis.
Loss of Wip1 activity coincides with the positivity of γH2AX during mitosis
Since we observed a decreased protein level of Wip1 in mitotic cells, we argued that also total enzymatic activity of Wip1 should be reduced during mitosis. To test this prediction, we established an assay for measuring the activity of endogenous Wip1 and indeed we found that the global Wip1 activity was decreased in mitotic cells (; Fig. S4). DDR kinases have recently been reported to support genome integrity by responding to and phosphorylating proteins at underreplicated DNA regions during unperturbed mitosis.Citation10,Citation11 According to this model, DNA lesions may accumulate in underreplicated DNA regions (such as chromosomal fragile sites) due to the mechanical forces acting during chromosome condensation or sister chromatid segregation.Citation10 These regions are marked by γH2AX during mitosis, followed by 53BP1 binding early in the G1 and protected throughout G1 until repair occurs in the following S phase.Citation10,Citation11 We and others have previously demonstrated that Wip1 dephosphorylates γH2AX during checkpoint recovery in interphase cells, thereby contributing to silencing of the DDR after successful DNA repair.Citation19-Citation21,Citation47 We hypothesized that removal of Wip1 during mitosis may contribute to the activation and impact of ATM and to the formation of γH2AX foci in mitotic cells. In good agreement with published data, we observed that mitotic cells are positive for γH2AX foci ().Citation10,Citation48,Citation49 Reduction of the number of mitotic γH2AX foci in the presence of selective ATMCitation50 and DNA-PKCitation51 (but not ATRCitation52) inhibitors suggests that both ATM and DNA-PK contribute to sensing of DNA damage in mitosis (Fig. S5). Importantly, overexpression of ectopic Wip1-WT impaired formation of γH2AX foci in mitotic cells (). Similarly, automated microscopic analysis revealed that expression of Wip1 (but not of a phosphatase dead Wip1-D314A) interfered with formation of 53BP1 nuclear bodies in G1 cells ().
Figure 4. Activity of Wip1 is decreased in mitosis. (A) Endogenous Wip1 was immunoprecipitated using rabbit anti-Wip1 (H300) from asynchronously growing or NZ-arrested cells and phosphatase activity was measured in vitro. (B) Expression of Wip1-WT was induced or not by tetracycline in asynchronously growing cells. Number of γH2AX positive foci was determined by a manual evaluation of immunofluorescence staining in 70 mitotic cells (n = 3, error bars indicate ,, ** p < 0.01). (C) Expression of Wip1 or Wip1-D314A was induced (+) or not (-) by tetracycline and cells were probed for 53BP1. Number of 53BP1 positive nuclear bodies in G1 cells was determined by automated microscopy in 1000 cells (n = 3, error bars indicate SD, ** p < 0.01). (D) Expression of Wip1-WT, Wip1–7A, Wip1–7D and Wip1-D314A was induced by tetracycline, FLAG-tagged proteins were immunoprecipitated and the phosphatase activity was determined in vitro (n = 3, error bars indicate SD, ** p < 0.01). (E) Expression of Wip1-WT, Wip1–7A, Wip1–7D and Wip1-D314A was induced (+) or not (-) by tetracycline and number of 53BP1 nuclear bodies was determined by automated microscopy. Intensity of DAPI signal was used for gating of G1 cells (n = 3, error bars indicate SD, ** p < 0.01). (F) Expression of Wip1-WT, Wip1–7A, Wip1–7D and Wip1-D314A was induced (+) or not (-) by tetracycline; cells were treated with 0.5 μM adriamycine for 4 h, and whole-cell lysates were probed for pSer15p53 and γH2AX.
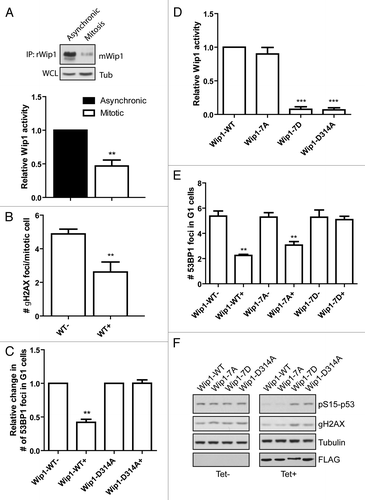
Furthermore, since we observed extensive mitotic phosphorylation of Wip1, we asked whether such modification might also contribute to the regulation of Wip1 enzymatic activity. Analysis of the activity of the modified Wip1-WT proved to be technically challenging, since immunopurified Wip1-WT showed a largely decreased mobility shift compared with Wip1-D314A, indicating that there is a considerable level of auto-dephosphorylation of overexpressed Wip1-WT during mitosis. Nevertheless, we observed a slightly decreased enzymatic activity of Wip1-WT purified from mitotic cells compared with that isolated from asynchronously growing cells (Fig. S6A). Next we investigated the enzymatic activity of Wip1 mutants. Whereas, there were no significant differences in the activities of Wip1-WT, Wip1-S40D and Wip1–7A, the activity of the phospho-mimicking mutant Wip1–7D was severely impaired (; Fig. S6B). In agreement with the differential enzymatic activity determined in vitro, induced expression of Wip1-WT or Wip1–7A (but not Wip1–7D) reduced the number of 53BP1 nuclear bodies in G1 cells (). In addition, Wip1-WT and Wip1–7A (but not Wip1–7D) impaired phosphorylation of the p53 tumor suppressor on Ser15 and γH2AX, both targets of the apical DDR kinases ATM and ATR, after exposure of cells to adriamycine (). From these experiments, we conclude that modification of a single Ser40 residue by Cdk1 during mitosis is not sufficient for inhibition of Wip1 activity but extensive modification of multiple residues within the catalytic domain impairs the activity of Wip1.
Overall, our data indicate that the observed decreased activity of Wip1 phosphatase during mitosis is ensured by coordinated complex phosphorylations of the Wip1 catalytic domain and decreased abundance of Wip1 protein due to enhanced proteolytic turnover.
Discussion
Whereas the roles of Wip1 in termination of signaling and in control of the recovery competence in response to genotoxic insults are well-established,Citation22,Citation25,Citation53,Citation54 molecular mechanisms that regulate Wip1 have remained largely unexplored. In this study, we investigated how human Wip1 phosphatase is regulated during the cell cycle. Here, we report that protein abundance of Wip1 increases from G1 to G2 and then rapidly declines in mitosis. The observation of differential expression of Wip1 during interphase was facilitated by development of models and techniques that allow analysis of live asynchronously growing cells, thus avoiding the need for cellular synchronization involving the treatment of cells with stress-inducing drugs (such as thymidine).Citation35,Citation59 These novel approaches include sorting of cells engineered to express fluorescent markers of the cell cycle progression and microscopic analysis of cells using intensity of the DAPI signal as the readout of the DNA content. Identification of higher levels of Wip1 in G2 compared with G1, reported here, is consistent with the fact that a complete loss of Wip1 in knockout mice resulted in an increase in the population of G2 cells.Citation55,Citation56 Wip1-knockout animals are viable, indicating that Wip1 is not essential for cell division.Citation56 Similarly, RNAi-mediated depletion of Wip1 in tissue culture cells caused a transient delay in G2/M transition, but eventually all cells enter mitosis.Citation25 Thus, Wip1 is not absolutely required for mitotic entry but might be involved in a tight control of the timing of the G2/M transition. Since expression of Wip1 is induced by various forms of genotoxic stress, it is plausible that higher levels of Wip1 observed in G2 may reflect the endogenous stress emanating from the unresolved replication intermediates of genomic DNA.Citation57 In turn, increased expression of Wip1 in G2 may timely counteract the ATR/Chk1 and ATM/Chk2 signaling modules that respond to replication stress and DNA breakage. In this way, the G2-associted elevation of Wip1 may facilitate cell cycle progression from G2 phase into mitosis.
In contrast to high levels in G2, abundance of Wip1 is dramatically decreased in early phases of mitosis. We have observed similar results using two antibodies recognizing distinct regions on Wip1 and ectopically expressed fluorescence-labeled Wip1, thereby excluding the possibility of epitope masking during mitosis. Given that the proteasomal inhibitor MG132 stabilized the mitotic Wip1 protein, and that no changes of Wip1 mRNA levels were detected throughout the cell cycle, we suggest that Wip1 is proteolytically degraded in mitosis. Whereas the degradation of Wip1 was apparently independent of SCF, depletion of the cdc20 caused stabilization of Wip1 in mitotic cells, indicating that Wip1 protein levels are controlled by the APC/Ccdc20 E3 ligase complex.
Induction of Wip1 expression using a tetracycline inducible system allowed us to investigate the phenotype of mitotic cells containing high levels of active Wip1 phosphatase. Since Wip1 can counteract the activity of ATM in response to DNA damage, and since ATM has recently been implicated in control of mitotic progression,Citation58 we tested the effect of Wip1 expression during mitosis. Although induction of Wip1-WT expression resulted in increased Wip1 activity in mitotic cells, we did not observe any major differences in the timing of mitotic progression, architecture of the mitotic spindle and cytokinesis (data not shown). In contrast, we observed that forced expression of Wip1-WT led to a dramatic decrease in the number of γH2AX foci in mitotic cells and also to a reduction of 53BP1 nuclear bodies in G1 cells. Since DNA lesions cannot be repaired during mitosis, it is essential to mark these regions by γH2AX to allow repair in the following cell cycle. Moreover, due to the lacking ubiquitination of mitotic chromatin, 53BP1 cannot bind to DNA lesions until cells exit from mitosis and enter the next G1.Citation12,Citation13 Thus, γH2AX appears to be an essential marker of endogenous DNA lesions present in unperturbed mitosis. Previously, we have shown that Wip1 associates with the chromatin and dephosphorylates γH2AX. Thus, it is likely that Wip1 activity needs to be eliminated from mitotic cells to allow formation of γH2AX foci at mitotic chromatin. Removing the phosphatase that has the ability to counteract the activity of ATM may allow cells to adequately respond to low levels of DNA damage present in unperturbed mitosis. Apart from the degradation of Wip1 during mitosis, we have also observed extensive phosphorylation of the catalytic domain of Wip1. Importantly, whereas the nonphosphorylatable Wip1–7A mutant protein showed normal enzymatic activity, the phosphomimicking Wip-7D mutant was enzymatically inactive. Therefore, we propose that cells efficiently reduce the net activity of Wip1 during mitosis through a combination of proteasomal degradation and phosphorylation-mediated inhibition of its enzymatic activity. After 53BP1 nuclear bodies form around and protect the DNA lesions in the following G1, Wip1 expression can increase again. In summary, we have shown here that protein abundance of Wip1 is not constant during the cell cycle but increases from G1 to G2, and then declines during mitosis. Degradation of Wip1 and inhibition of Wip1 enzymatic activity by extensive post-translational modifications at multiple residues within the catalytic domain of Wip1 in mitosis, allow more sensitive DNA damage signaling during mitosis, including appearance of γH2AX foci at mitotic chromatin. Such cell cycle-regulated modulation of the DDR signaling threshold through changes in Wip1 abundance and activity seem to facilitate formation of the 53BP1 nuclear (OPT) bodiesCitation10,Citation11 in the subsequent G1 phase, thereby contributing to the ability of mammalian cells to adequately deal with consequences of replication stress and, indeed, to the global maintenance of genome integrity.
Materials and Methods
Antibodies and reagents
The following antibodies were used in this study: rabbit Wip1 (H300; sc20712), mouse Wip1 (clone F-10; sc37625), p53 (sc-6243), cyclin A (sc-53230), cdc20 (sc-8358) and 53BP1 (sc-22760; Santa Cruz Biotechnology); Wip1/PPM1D (A300–664A, Bethyl Laboratories), pSer15p53 (#9284S), β-TrCP (#4394), cyclin B1 (V152; #4135), γH2AX (#2577; Cell Signaling Technology); α-tubulin (T5168) and FLAG (F1804; Sigma Aldrich); Cyclin B (ab7957; Abcam); pSer10-H3 (06–570; Millipore-Upstate); GAPDH (GTX30666; GeneTex); cdc27 (#610454, BD Transduction Laboratories), Alexa Fluor fluorescently labeled secondary antibodies were from Life Technologies. Lambda phosphatase was from New England Biolabs. PhosTag acrylamide was from Wako Pure Chemical Industries (AAL-107).
Plasmids
pRS-Wip1 construct targeting GGATGACTTTGTCAGAGCT of the human Wip1, FLAG tagged pcdna4/TO-Wip1-WT, pcdna4/TO-Wip1-D314A and pWip1-mCherry were described previously.Citation19,Citation25 Mutagenesis was performed using QuickChange II site-directed mutagenesis kit (Agilent Technologies), and fragments encoding Wip1-S40A, S40D, T410A-S412A-S416A (Wip1-AAA), S97A, S97D were inserted into pcdna4/TO (Life Technologies) or pQE81L (Qiagen). Mutants Wip1–7A (containing T34/S40/S44/S46/S54/S85/S97A) and Wip1–7D (containing T34/S40/S44/S46/S54/S85/ S97D) were generated by GenArt Synthesis (Life Technologies), and fragments were inserted into pcdna4/TO. pFucci-S/G2/M-Green and pFucci-G1 orange plasmids expressing hGeminin (1–110) and hCdt1 (30–120) fragments fused with fluorescent reporters are based on published fluorescent ubiquitination-based cell cycle indicator (FUCCI) and were from MBL International.Citation36
Cell lines
Human HeLa, MCF7 and U2OS cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) under standard conditions as described.Citation59 Human hTERT-immortalized retinal pigment epithelial cells (RPE) were grown in DMEM supplemented with nutrient mixture F-12 (DMEM/F12). Cells expressing Wip1, or its point mutants, under control of tetracycline-inducible promoter were generated as describedCitation25 and routinely grown in media containing Tet system-approved serum.Citation25 Enhanced expression was induced by addition of tetracycline (1 µg/ml) for 12 h. Monoclonal cell line stably expressing FUCCI indicator was generated by transfection of U2OS cells followed by hygromycine/neomycine selection and two steps of clonal selection using limited dilution on a 96-well plate. Live cells positive for GFP (corresponding to G2 cells) or double-negative cells (DN) and RFP-positive cells (corresponding to G1 cells) were sorted using Influx cell sorter (BD Biosciences) and lysed for biochemical analysis. In parallel, cells pre-incubated with a cell permeable dye Hoechst 33342 as a DNA marker were analyzed by flow cytometry.
Cell synchronization
Cells were synchronized at G1/S transition using a double thymidine (2.5 mM) block (12 h-Thy/8 h-release/12 h-Thy), washed in PBS and released into fresh media supplemented with nocodazole to arrest cells in mitosis (most cells entered M after 10–12 h). Cells were collected in 2-h intervals. Mitotic cells were collected by shake-off. Alternatively, cells were synchronized at late G2 using Ro-3306 (9 μM for 20 h; Tocris Bioscience) and released into fresh media (most cells entered M after 1–2 h). Where indicated, MG132 (5 μM, Sigma) was added to media to block proteasomal degradation.
Immunofluorescence and live-cell imaging
Cells were grown in 96-well imaging plates (BD Biosciences) and immunofluorescence was performed as described.Citation60 For each image, the background signal of an area without cells was subtracted. Nuclei were identified based on DAPI staining before measurement of nuclear Wip1 staining. Images were acquired on a DeltaVision Spectris imaging system equipped with a 20× objective, NA 0.7 (Applied Precision). For automated microscopic analysis of 53BP1 or γH2AX-positive foci, cells were cultured on glass coverslips, fixed using 4% formaldehyde (10 min at RT), permeabilized by ice-cold methanol, blocked with 3% BSA in PBS supplemented with 0.1% Tween-20 (PBST) and incubated with the primary 53BP1 or γH2AX antibodies for 1 h at RT. After washing with PBST, coverslips were incubated with AlexaFluor-conjugated secondary antibodies for 1 h at RT. DNA was stained with DAPI (Sigma) for 2 min and coverslips were mounted using Vectashield reagent (Vector Laboratories). Automated image acquisition was done using Scan^R high-content screening station equipped with a motorized stage and 40× objective (Olympus). Nuclei were identified based on the DAPI signal and the median number of 53BP1 or γH2AX foci was determined using a spot detection module. Mitotic cells were gated based on higher DAPI intensity of condensed chromatin with manual correction for false-positive events. At least 1,000 nuclei for interphase cells and at least 100 mitotic cells were counted per condition in three independent experiments. For live-cell imaging cells expressing FUCCI, indicator were grown in LabTek II chamber slides in CO2-independent L15 media (Gibco) and imaged at 30 min intervals using Leica DMI6000 inverted microscope.
Transfection
U2OS cells were transfected with plasmid DNA using a standard calcium phosphate method. Silencer Select siRNAs targeting Cul1 (GGCUUGUGGUCGCUUCAU), βTrCP1 (GAACUAUAAA GGUAUGGAA), βTrCP2 (GGUUGUUAGUGGAUCAUCA), Fbx7 (GGGUUGUUAGUGGAGCAUA) and cdc20 (CGAAAUGACUAUUACCUGA) were obtained from Ambion (Life Technologies) and were transfected at 5 nM concentration using RNAiMAX (Life Technologies) and collected at indicated times post-transfection.
In vitro phosphatase assay
Cells were induced by tetracycline to express FLAG-tagged Wip1-Wt or its point mutants, extracted by EBC buffer (50 mM Tris pH7.5, 150 mM NaCl, 0.5% NP-40, 1 mM EGTA) and subsequent sonication (3 × 10 sec, amplitude 7, 100 W) and extracts were spinned down (16.000 g, 10 min, 4°C) and normalized for protein concentration. FLAG-tagged proteins were immunopurified from tetracycline-induced cells with anti-FLAG agarose (Sigma). Alternatively, endogenous Wip1 was immunoprecipitated using a polyclonal anti-Wip1 antibody (H300, 2 μg/reaction) and 30 μl protein A/G Ultra link resin (Thermo Scientific). Phosphatase assay was performed using synthetic phosphopeptide, corresponding to pSer15p53 (100 μM, GeneScript) and Biomol Green reagent (Enzo Life Sciences) as described.Citation19 Statistical analysis was performed using Student’s t-test in GraphPad Prism software.
In vitro kinase assay
E. coli-(BL21) transformed with pQE81L-Wip1-D314A or its point mutants were induced with 0.5 mM IPTG for 4 h at 37°C and His-tagged Wip1 was purified using Ni-NTA agarose (Qiagen). His-Wip1 (2 mg) was mixed with 50 ng of active protein kinases Cdk1/cyclin B (Sigma) in kinase buffer (25 mM MOPS pH 7.2, 12.5 mM glycerol 2-phosphate, 25 mM MgCl2, 5 mM EGTA, 2 mM EDTA and 0.25 mM DTT) supplemented with 100 µM ATP and 5 µCi 32P-γ-ATP and a sample was incubated for 20 min at 30°C. Proteins were separated using SDS-PAGE as described,Citation61 and phosphorylation was detected by autoradiography.
Quantitative real-time PCR (qPCR)
Total RNA samples were isolated using the TRI Reagent (Sigma) according to the manufacturer’s protocol. First-strand cDNA was synthesized from 200 ng of total RNA with random hexamer primers using TaqMan Reverse Transcription Reagents (Applied Biosystems). qRT-PCR was performed in ABI Prism 7300 (Applied Biosystems) using SYBR Green I Master Mix with the following set of primers: Wip1 (GGGAGTGATGGACTTTGGAA and CAAGATTGTCCATGCTCACC), tubulin (GAGTGCATCTCCATCCACGTT and TAGAGCTCCCAGCAGGCATT). The relative quantity of cDNA was estimated by ΔΔCt, and data were normalized to tubulin. Samples were measured in triplicate in three independent experiments.
Mass spectrometry
Phosphatase dead FLAG-Wip1-D314A was immunoprecipitated from mitotic cells obtained by mitotic shake-off of nocodazole-arrested cells. The sample was separated on SDS-PAGE, stained by CBB and a slice of the gel corresponding to the Wip1 was digested by trypsin or V8 protease and peptides were subjected to MS. Peptide mass was analyzed using Mascot software (Matrix Science). Combined sequence coverage was 82%.
| Abbreviations: | ||
| 53BP1 | = | p53-binding protein 1 |
| APC/C | = | anaphase-promoting complex/cyclosome |
| ATM | = | ataxia telangiectasia-mutated kinase |
| ATR | = | Ataxia telangiectasia and Rad3-related kinase |
| BRCA1 | = | breast cancer type 1 susceptibility protein |
| Cdk | = | cyclin-dependent kinase |
| DDR | = | DNA damage response |
| FUCCI | = | fluorescent, ubiquitination-based cell cycle indicator |
| γH2AX | = | histone variant H2AX, phosphorylated on serine 139 |
| IR | = | ionizing radiation |
| MDC1 | = | mediator of DNA damage checkpoint protein 1 |
| NZ | = | nocodazole |
| PPM1D | = | Protein phosphatase Mg2+/Mn2+ dependent 1D |
| SCF | = | Skp, Cullin, F-box containing complex |
| WCL | = | whole-cell lysate |
| Wip1 | = | wild-type p53 induced phosphatase 1 |
Additional material
Download Zip (790.4 KB)Acknowledgments
We are thankful to Dr. Staňek (IMG) for making the Scan^R imaging station available and all members of our laboratories for helpful discussions. This work was supported by the Grant Agency of the Czech Republic (projects P305/10/P420 and P301/10/1525), the Netherlands Genomic Initiative of NWO (CGC), the Danish Cancer Society and the European Commission (project DDResponse), the Swedish research council (A.L.), the Swedish cancer foundation (A.L.) and the Swedish childhood cancer foundation (A.L.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003; 3:155 - 68; http://dx.doi.org/10.1038/nrc1011; PMID: 12612651
- Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol 2011; 13:1161 - 9; http://dx.doi.org/10.1038/ncb2344; PMID: 21968989
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005; 123:1213 - 26; http://dx.doi.org/10.1016/j.cell.2005.09.038; PMID: 16377563
- Huen MSY, Grant R, Manke I, Minn K, Yu X, Yaffe MB, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007; 131:901 - 14; http://dx.doi.org/10.1016/j.cell.2007.09.041; PMID: 18001825
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007; 131:887 - 900; http://dx.doi.org/10.1016/j.cell.2007.09.040; PMID: 18001824
- Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009; 136:435 - 46; http://dx.doi.org/10.1016/j.cell.2008.12.041; PMID: 19203579
- Bekker-Jensen S, Rendtlew Danielsen J, Fugger K, Gromova I, Nerstedt A, Lukas C, et al. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat Cell Biol 2010; 12:80 - 6, 1-12; http://dx.doi.org/10.1038/ncb2008; PMID: 20023648
- Moudry P, Lukas C, Macurek L, Hanzlikova H, Hodny Z, Lukas J, et al. Ubiquitin-activating enzyme UBA1 is required for cellular response to DNA damage. Cell Cycle 2012; 11:1573 - 82; http://dx.doi.org/10.4161/cc.19978; PMID: 22456334
- Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 2012; 47:497 - 510; http://dx.doi.org/10.1016/j.molcel.2012.07.029; PMID: 22920291
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 2011; 13:243 - 53; http://dx.doi.org/10.1038/ncb2201; PMID: 21317883
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, et al. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol 2011; 193:97 - 108; http://dx.doi.org/10.1083/jcb.201011083; PMID: 21444690
- Giunta S, Belotserkovskaya R, Jackson SP. DNA damage signaling in response to double-strand breaks during mitosis. J Cell Biol 2010; 190:197 - 207; http://dx.doi.org/10.1083/jcb.200911156; PMID: 20660628
- Giunta S, Jackson SP. Give me a break, but not in mitosis: the mitotic DNA damage response marks DNA double-strand breaks with early signaling events. Cell Cycle 2011; 10:1215 - 21; http://dx.doi.org/10.4161/cc.10.8.15334; PMID: 21412056
- Harris DR, Bunz F. Protein phosphatases and the dynamics of the DNA damage response. Cell Cycle 2010; 9:861 - 9; http://dx.doi.org/10.4161/cc.9.5.10862; PMID: 20348842
- Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell 2006; 23:757 - 64; http://dx.doi.org/10.1016/j.molcel.2006.07.010; PMID: 16949371
- Fujimoto H, Onishi N, Kato N, Takekawa M, Xu XZ, Kosugi A, et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ 2006; 13:1170 - 80; http://dx.doi.org/10.1038/sj.cdd.4401801; PMID: 16311512
- Lu X, Ma O, Nguyen T-A, Jones SN, Oren M, Donehower LA. The Wip1 Phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell 2007; 12:342 - 54; http://dx.doi.org/10.1016/j.ccr.2007.08.033; PMID: 17936559
- Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev 2005; 19:1162 - 74; http://dx.doi.org/10.1101/gad.1291305; PMID: 15870257
- Macůrek L, Lindqvist A, Voets O, Kool J, Vos HR, Medema RH. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene 2010; 29:2281 - 91; http://dx.doi.org/10.1038/onc.2009.501; PMID: 20101220
- Moon S, Lin L, Zhang X, Nguyen T, Darlington Y, Waldman A. Wildtype p53-induced phosphatase 1 dephosphorylates histone variant {gamma}-H2AX and suppresses DNA double strand break repair. J Biol Chem 2010; 23:12935 - 47; http://dx.doi.org/10.1074/jbc.M109.071696
- Cha H, Lowe JM, Li H, Lee J-S, Belova GI, Bulavin DV, et al. Wip1 directly dephosphorylates γ-H2AX and attenuates the DNA damage response. Cancer Res 2010; 70:4112 - 22; http://dx.doi.org/10.1158/0008-5472.CAN-09-4244; PMID: 20460517
- Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, Donehower LA. The type 2C phosphatase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev 2008; 27:123 - 35; http://dx.doi.org/10.1007/s10555-008-9127-x; PMID: 18265945
- Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci USA 1997; 94:6048 - 53; http://dx.doi.org/10.1073/pnas.94.12.6048; PMID: 9177166
- Zhang X, Wan G, Mlotshwa S, Vance V, Berger FG, Chen H, et al. Oncogenic Wip1 phosphatase is inhibited by miR-16 in the DNA damage signaling pathway. Cancer Res 2010; 70:7176 - 86; http://dx.doi.org/10.1158/0008-5472.CAN-10-0697; PMID: 20668064
- Lindqvist A, de Bruijn M, Macurek L, Brás A, Mensinga A, Bruinsma W, et al. Wip1 confers G2 checkpoint recovery competence by counteracting p53-dependent transcriptional repression. EMBO J 2009; 28:3196 - 206; http://dx.doi.org/10.1038/emboj.2009.246; PMID: 19713933
- Nannenga B, Lu X, Dumble M, Van Maanen M, Nguyen T-A, Sutton R, et al. Augmented cancer resistance and DNA damage response phenotypes in PPM1D null mice. Mol Carcinog 2006; 45:594 - 604; http://dx.doi.org/10.1002/mc.20195; PMID: 16652371
- Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet 2004; 36:343 - 50; http://dx.doi.org/10.1038/ng1317; PMID: 14991053
- Shreeram S, Hee WK, Demidov ON, Kek C, Yamaguchi H, Fornace AJ Jr., et al. Regulation of ATM/p53-dependent suppression of myc-induced lymphomas by Wip1 phosphatase. J Exp Med 2006; 203:2793 - 9; http://dx.doi.org/10.1084/jem.20061563; PMID: 17158963
- Rauta J, Alarmo E-L, Kauraniemi P, Karhu R, Kuukasjärvi T, Kallioniemi A. The serine-threonine protein phosphatase PPM1D is frequently activated through amplification in aggressive primary breast tumours. Breast Cancer Res Treat 2006; 95:257 - 63; http://dx.doi.org/10.1007/s10549-005-9017-7; PMID: 16254685
- Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet 2002; 31:210 - 5; http://dx.doi.org/10.1038/ng894; PMID: 12021785
- Li J, Yang Y, Peng Y, Austin RJ, van Eyndhoven WG, Nguyen KCQ, et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat Genet 2002; 31:133 - 4; http://dx.doi.org/10.1038/ng888; PMID: 12021784
- Castellino RC, De Bortoli M, Lu X, Moon S-H, Nguyen T-A, Shepard MA, et al. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J Neurooncol 2008; 86:245 - 56; http://dx.doi.org/10.1007/s11060-007-9470-8; PMID: 17932621
- Saito-Ohara F, Imoto I, Inoue J, Hosoi H, Nakagawara A, Sugimoto T, et al. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res 2003; 63:1876 - 83; PMID: 12702577
- Tan DSP, Lambros MBK, Rayter S, Natrajan R, Vatcheva R, Gao Q, et al. PPM1D is a potential therapeutic target in ovarian clear cell carcinomas. Clin Cancer Res 2009; 15:2269 - 80; http://dx.doi.org/10.1158/1078-0432.CCR-08-2403; PMID: 19293255
- Bolderson E, Scorah J, Helleday T, Smythe C, Meuth M. ATM is required for the cellular response to thymidine induced replication fork stress. Hum Mol Genet 2004; 13:2937 - 45; http://dx.doi.org/10.1093/hmg/ddh316; PMID: 15459181
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008; 132:487 - 98; http://dx.doi.org/10.1016/j.cell.2007.12.033; PMID: 18267078
- Song JY, Han H-S, Sabapathy K, Lee B-M, Yu E, Choi J. Expression of a homeostatic regulator, Wip1 (wild-type p53-induced phosphatase), is temporally induced by c-Jun and p53 in response to UV irradiation. J Biol Chem 2010; 285:9067 - 76; http://dx.doi.org/10.1074/jbc.M109.070003; PMID: 20093361
- Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, et al. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci USA 2006; 103:10660 - 5; http://dx.doi.org/10.1073/pnas.0600447103; PMID: 16818887
- Skaar JR, Pagano M. Control of cell growth by the SCF and APC/C ubiquitin ligases. Curr Opin Cell Biol 2009; 21:816 - 24; http://dx.doi.org/10.1016/j.ceb.2009.08.004; PMID: 19775879
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001; 294:173 - 7; http://dx.doi.org/10.1126/science.1065203; PMID: 11533444
- Chan EH, Santamaria A, Silljé HH, Nigg EA. Plk1 regulates mitotic Aurora A function through betaTrCP-dependent degradation of hBora. Chromosoma 2008; 117:457 - 69; http://dx.doi.org/10.1007/s00412-008-0165-5; PMID: 18521620
- Wolthuis R, Clay-Farrace L, van Zon W, Yekezare M, Koop L, Ogink J, et al. Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol Cell 2008; 30:290 - 302; http://dx.doi.org/10.1016/j.molcel.2008.02.027; PMID: 18471975
- den Elzen N, Pines J. Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J Cell Biol 2001; 153:121 - 36; http://dx.doi.org/10.1083/jcb.153.1.121; PMID: 11285279
- Geley S, Kramer E, Gieffers C, Gannon J, Peters J-M, Hunt T. Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J Cell Biol 2001; 153:137 - 48; http://dx.doi.org/10.1083/jcb.153.1.137; PMID: 11285280
- Yamada S, Nakamura H, Kinoshita E, Kinoshita-Kikuta E, Koike T, Shiro Y. Separation of a phosphorylated histidine protein using phosphate affinity polyacrylamide gel electrophoresis. Anal Biochem 2007; 360:160 - 2; http://dx.doi.org/10.1016/j.ab.2006.10.005; PMID: 17092477
- Dinkel H, Michael S, Weatheritt RJ, Davey NE, Van Roey K, Altenberg B, et al. ELM--the database of eukaryotic linear motifs. Nucleic Acids Res 2012; 40:Database issue D242 - 51; http://dx.doi.org/10.1093/nar/gkr1064; PMID: 22110040
- Moon S-H, Nguyen T-A, Darlington Y, Lu X, Donehower LA. Dephosphorylation of γ-H2AX by WIP1: an important homeostatic regulatory event in DNA repair and cell cycle control. Cell Cycle 2010; 9:2092 - 6; http://dx.doi.org/10.4161/cc.9.11.11810; PMID: 20495376
- Ichijima Y, Sakasai R, Okita N, Asahina K, Mizutani S, Teraoka H. Phosphorylation of histone H2AX at M phase in human cells without DNA damage response. Biochem Biophys Res Commun 2005; 336:807 - 12; http://dx.doi.org/10.1016/j.bbrc.2005.08.164; PMID: 16153602
- McManus KJ, Hendzel MJ. ATM-dependent DNA damage-independent mitotic phosphorylation of H2AX in normally growing mammalian cells. Mol Biol Cell 2005; 16:5013 - 25; http://dx.doi.org/10.1091/mbc.E05-01-0065; PMID: 16030261
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NMB, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 2004; 64:9152 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-04-2727; PMID: 15604286
- Leahy JJJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, et al. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett 2004; 14:6083 - 7; http://dx.doi.org/10.1016/j.bmcl.2004.09.060; PMID: 15546735
- Reaper PM, Griffiths MR, Long JM, Charrier J-D, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011; 7:428 - 30; http://dx.doi.org/10.1038/nchembio.573; PMID: 21490603
- Le Guezennec X, Bulavin DV. WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem Sci 2010; 35:109 - 14; http://dx.doi.org/10.1016/j.tibs.2009.09.005; PMID: 19879149
- Lu X, Nguyen T-A, Donehower LA. Reversal of the ATM/ATR-mediated DNA damage response by the oncogenic phosphatase PPM1D. Cell Cycle 2005; 4:1060 - 4; http://dx.doi.org/10.4161/cc.4.8.1876; PMID: 15970689
- Zhu Y-H, Zhang C-W, Lu L, Demidov ON, Sun L, Yang L, et al. Wip1 regulates the generation of new neural cells in the adult olfactory bulb through p53-dependent cell cycle control. Stem Cells 2009; 27:1433 - 42; http://dx.doi.org/10.1002/stem.65; PMID: 19489034
- Choi J, Nannenga B, Demidov ON, Bulavin DV, Cooney A, Brayton C, et al. Mice deficient for the wild-type p53-induced phosphatase gene (Wip1) exhibit defects in reproductive organs, immune function, and cell cycle control. Mol Cell Biol 2002; 22:1094 - 105; http://dx.doi.org/10.1128/MCB.22.4.1094-1105.2002; PMID: 11809801
- Petermann E, Caldecott KW. Evidence that the ATR/Chk1 pathway maintains normal replication fork progression during unperturbed S phase. Cell Cycle 2006; 5:2203 - 9; http://dx.doi.org/10.4161/cc.5.19.3256; PMID: 16969104
- Yang C, Tang X, Guo X, Niikura Y, Kitagawa K, Cui K, et al. Aurora-B mediated ATM serine 1403 phosphorylation is required for mitotic ATM activation and the spindle checkpoint. Mol Cell 2011; 44:597 - 608; http://dx.doi.org/10.1016/j.molcel.2011.09.016; PMID: 22099307
- Hubackova S, Novakova Z, Krejcikova K, Kosar M, Dobrovolna J, Duskova P, et al. Regulation of the PML tumor suppressor in drug-induced senescence of human normal and cancer cells by JAK/STAT-mediated signaling. Cell Cycle 2010; 9:3085 - 99; http://dx.doi.org/10.4161/cc.9.15.12521; PMID: 20699642
- Lindqvist A, van Zon W, Karlsson Rosenthal C, Wolthuis RMF. Cyclin B1-Cdk1 activation continues after centrosome separation to control mitotic progression. PLoS Biol 2007; 5:e123; http://dx.doi.org/10.1371/journal.pbio.0050123; PMID: 17472438
- Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, Bartek J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a). Cell Cycle 2011; 10:457 - 68; http://dx.doi.org/10.4161/cc.10.3.14707; PMID: 21248468