Recent advances in cancer research has revealed that tumors should be considered as an integrated network of neoplastic and ancillary cells of the tumor microenvironment, including cancer- associated fibroblasts (CAFs), macrophages, endothelial cells, etc.Citation1 The cross-talk between these populations is not only merely mediated by soluble cytokines and growth factors, but is now been enlarged to metabolites, like lactate, ketone bodies or proteins.Citation2,Citation3 These metabolites are exchanged among stromal and cancer cells or using membrane solute transporters, or mediated by release of cargo-vesicles, such as exosomes or oncosomes, which can carry ATP, proteins or miRNAs.Citation4
This intra-tumoral cross-talk elicits, in cancer cells, an escaping strategy called epithelial mesenchymal transition (EMT) accompanied by expression of stem-like traits and granting for successful metastases.Citation5 In addition, stromal and cancer cells undergo a reciprocal metabolic reprogramming, useful to sustain cancer cells survival and growth. In particular, in stromal cells, cancer cells induce a Warburg-like metabolism, fueling cancer cells themselves with essential metabolites such as lactate and ketone bodies. Cancer cells use these energy-rich molecules for anabolic purposes as well as to fuel ATP synthesis through respiration, undergoing the so-called “reverse Warburg” metabolism.Citation3,Citation6
This coupling metabolism within tumor stroma, involving direct and reverse Warburg metabolism, could explain the controversial data concerning the role of mitochondria in cancer progression. Indeed, several data indicate a mandatory role of mitochondria in cancer cells, ranging from lactate respiration, to Krebs cycle fueling with ketone bodies, to citrate exportation to fuel fatty acids synthesis.Citation7 In addition, mitochondrial metabolism is compulsory for glutamine-addicted cancer cells. The most likely hypothesis is that in cancer cells, mitochondrial reprogramming leads to a shift toward ketone/glutamine utilization, leading to citrate-mediated fatty acids synthesis. This integrated behavior commits stromal cells to a less efficient metabolism (extrusion of energy-rich metabolites and mitochondrial-independent energy production), and cancer cells to exploit stromal cells to fulfill their survival and growth in ischemic environment. This interplay presumes that cancer cells rely extensively on mitochondrial functions.
The paper from Lisanti’s groupCitation8 perfectly fits with this scenario in a breast cancer model, as it points out that the effects of inhibition of mitochondrial function are different between cancer and stromal cells, thereby giving an explanation to the antiproliferative effects of metformin. The latter is a widely used oral antidiabetic drug, endowed with promising effects for cancer prevention and treatment.Citation9 Metformin is reported to inhibit mitochondrial complex I activity, thereby disrupting oxidative mitochondrial metabolism, mandatory for cancer reverse Warburg metabolism and granting survival and growth. Sanchez-Alvarez and colleagues,Citation8 although not directly using metformin, reported that uncoupling protein-mediated mitochondrial dysfunction actually has compartment-specific effects. Indeed, mitochondrial dysfunction in stromal CAFs enhances their metabolic reprogramming to production of energy-rich metabolites, increasing tumor growth. On the contrary, disruption of mitochondrial function through UCP overexpression in cancer cells leads to the opposite effect, restraining tumor growth.
The outcome of these data are that, in vivo, mitochondrial functional disruption may have positive or negative effects on cancer progression, depending on the effective addiction of different cancers from their stromal counterparts. A latere, with respect to the epidemiologic data on diabetic patients treated with metformin, showing a reduced risk of cancer onset, the final answer to the question on in vivo effects of mitochondrial drugs for cancer progression will arise from the several ongoing clinical trials assessing the effect of adding metformin to the existing chemotherapy regimen in the treatment of cancers ().
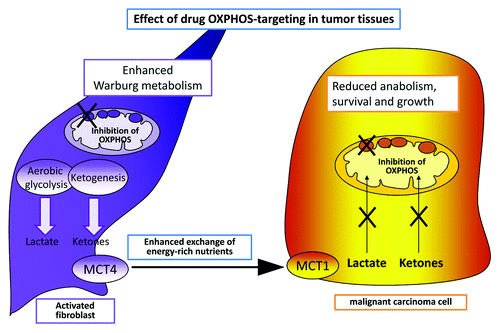
Figure 1. Targeting OXPHOS in tumor microenvironment. Inhibition of OXPHOS in the whole tumor tissue gives rise to opposite effects in tumor cells or in stromal counterparts. Indeed, the block of mitochondrial machinery in CAFs activate a Warburg-like metabolism, forcing the production of lactate and ketones, energy-rich metabolites that cancer cells are no more able to utilize for anabolic purpose due to the block of their own mitochondrial metabolism by drugs. In addition, during OXPHOS targeting of tumors, accumulating lactate and ketones likely participate to acidify the hostile tumor microenvironment.
References
- Cirri P, et al. Cancer Metastasis Rev 2012; 31:195 - 208; http://dx.doi.org/10.1007/s10555-011-9340-x; PMID: 22101652
- Bonuccelli G, et al. Cell Cycle 2010; 9:3506 - 14; http://dx.doi.org/10.4161/cc.9.17.12731; PMID: 20818174
- Fiaschi T, et al. Cancer Res 2012; 72:5130 - 40; http://dx.doi.org/10.1158/0008-5472.CAN-12-1949; PMID: 22850421
- Bobrie A, et al. Cancer Res 2012; 72:4920 - 30; http://dx.doi.org/10.1158/0008-5472.CAN-12-0925; PMID: 22865453
- Giannoni E, et al. Cancer Res 2010; 70:6945 - 56; http://dx.doi.org/10.1158/0008-5472.CAN-10-0785; PMID: 20699369
- Pavlides S, et al. Cell Cycle 2009; 8:3984 - 4001; http://dx.doi.org/10.4161/cc.8.23.10238; PMID: 19923890
- Moreno-Sánchez R, et al. FEBS J 2007; 274:1393 - 418; http://dx.doi.org/10.1111/j.1742-4658.2007.05686.x; PMID: 17302740
- Sanchez-Alvarez R, et al. Cell Cycle 2013; 12; In this issue http://dx.doi.org/10.4161/cc.23058
- Bost F, et al. Curr Opin Oncol 2012; 24:103 - 8; http://dx.doi.org/10.1097/CCO.0b013e32834d8155; PMID: 22123231