Abstract
The induction of senescence in tumor cells impairs transformation and promotes an anticancer immune response resulting from the production by senescent cells of cytokines and chemokines, an aspect known as “senescence-associated secretory phenotype” (SASP). Here we discuss recent findings regarding the role of NFκB in the modulation of the SASP and the consequent anticancer immune response.
Keywords: :
Cellular senescence constitutes an irreversible cell cycle arrest that occurs in normal cells in response to various adverse conditions, including telomere shortening in replicative senescenceCitation1 as well as telomere dysfunction in oncogene-induced senescence (OIS).Citation2 In addition, premature (or accelerated) senescence can be readily elicited in tumor cells by the administration of sublethal concentrations of conventional anticancer drugs.Citation3
The acquisition of a senescent phenotype in cells from different tissues (e.g., fibroblast, epithelial or endothelial cells) is accompanied by an increased secretion of specific cytokines, chemokines and other factors, which affect both senescent and neighboring, non-senescent cells via autocrine/paracrine mechanism. The secretion of these factors has been referred to as “senescence-associated secretory phenotype” (SASP).Citation4 Notably, the SASP occurs not only in vitro, but also in vivo, in response to oncogene activation or DNA-damaging chemotherapy.Citation5
The induction of cell senescence has been associated with various anticancer effects. For instance, OIS acts as a very effective barrier to cell transformation, in both mouse models and in human cancers.Citation6 In addition, the induction of an accelerated senescent phenotype by chemotherapeutic agents critically contributes to the efficacy of anticancer therapy.Citation7 The SASP has been implicated in several of the tumor suppressive effects of senescence. Kuilman and colleagues identified interleukin (IL)-6 and IL-8 as essential mediators of oncogenic BRAF-induced senescence.Citation8 In response to oncogenic stress, the activation of the transcription factor C/EBPβ stimulates IL-6 and IL-8 secretion. Interference with either of these cytokines, or with their receptors, resulted in the bypass of the senescence. Interestingly, IL-6 depletion also reduced C/EBPβ levels, highlighting the existence of a positive feedback loop that acts to sustain the senescence-associated cell cycle arrest. Membrane bound IL-1α has also been shown to act as a positive regulator of IL-6 and IL-8 expression in senescent human fibroblasts.Citation9 Indeed, the autocrine stimulation of senescent cells with IL-1α induces the constitutive activation of NFκB, which in turn transactivates IL-1α, resulting in another positive feedback loop. The inhibition of IL-1α signaling limited both NFκB and C/EBPβ activity, as well as the secretion of IL-6 and IL-8.Citation9 Finally, a positive feedback loop involving the chemokine receptor CXCR2 and its ligands, namely IL-8 and GRO-α, has been shown to act to reinforce replicative senescence in primary human fibroblasts.Citation10 NFκB and C/EBPβ coordinately regulate the expression of CXCR2 ligands during OIS.Citation10 Hence, several components involved in the SASP operate within positive feedback loops that reinforce the growth arrest associated with cell senescence.
Besides contributing to the autocrine loops that reinforce the senescent phenotype, the SASP is also responsible—at least in part—for a crosstalk between senescent cells and cells of the immune system. This crosstalk mediates additional anticancer function of senescence, as it is important for the recruitment of immune cells to the tumor site as well as for clearance of senescent cells. Such an immunomodulatory function of the SASP has been described in normal senescent cellsCitation11 as well as in senescent tumor cells.Citation12,Citation13 For instance, the restoration of endogenous p53 expression in hepatocarcinoma and sarcoma leads to induction of a senescence program that triggers tumor clearance in vivo, through the activation of an innate immune response. The molecular mechanisms mediating such anticancer immune responses have not yet been fully elucidated, although SASP components including several immune modulators are likely to be involved.Citation4,Citation11 In addition, it has been shown that senescent myeloma cells express natural killer (NK) cell-activating ligands on their surface.Citation14
We recently investigated the crosstalk between prematurely senescent tumor cells and cells of the innate immune system, as well as the potential involvement of death receptors in the recognition and clearance of premature senescent carcinoma cells by the immune system.Citation15 We evaluated the expression of FAS, DR4 and DR5 on proliferating and senescent (in response to chemotherapy) induced lung adenocarcinoma A549 and breast carcinoma MCF7 cells. We showed that the induction of senescence in both tumor cell lines is associated with an increase in FAS expression. Such upregulation of FAS in senescent cells sensitized cells to the induction of apoptosis.
Inflammatory cytokines have previously been implicated in the modulation of FAS expression in different cell types. For instance, the administration of both tumor necrosis factor α (TNFα) and interferon γ (IFNγ) has been shown to trigger FAS upregulation in murine fibroblasts.Citation16 IFNγ combined with IL-1β also increased FAS expression on human thyrocytes, resulting in enhanced susceptibility to FAS ligand (FASL)-mediated cell death.Citation17 Finally, human endometrial stromal cells are normally resistant to FAS-dependent apoptosis, but become readily sensitive upon the administration of TNFα and IFNγ, which cause FAS upregulation.Citation18 These observations prompted us to investigate whether increased FAS expression in senescent tumor cells was dependent on the SASP. Analyses of media conditioned by senescent MCF-7 cells highlighted an increased secretion of several cytokines and chemokines.Citation15 Among these cytokines, we identified TNFα and IFNγ as mediating the upregulation of FAS in our experimental system. Accordingly, the treatment of proliferating cancer cell lines with TNFα and IFNγ resulted in FAS upregulation, while interfering with the TNFα and IFNγ signaling pathways decreased the amount of FAS expressed on the surface senescent tumor cells. Hence, SASP components are likely to promote the recognition of senescent carcinoma cells and their elimination by FASL-positive immune cells.
Previous studies have demonstrated a crucial role for NFκB in the induction of FAS expression.Citation16,Citation19 Since an increased activity of NFκB has been associated with cellular senescence,Citation10 we investigated the ability of NFκB to modulate the SASP in carcinoma cells pushed into senescence by the administration of chemotherapeutic drugs. To this aim, we knocked-down the expression of RelA, the main member of the NFκB family, in MCF-7 and A549 cells by RNA interference, and then analyzed media conditioned from such cells (or cells receiving a scamble siRNA) upon the induction of senescence. Interestingly, many SASP components were significantly reduced in RelA-depleted cells.Citation15 The essential role of NFκB in modulating the SASP has recently been substantiated in response to diverse senescence-inducing stimuli.Citation20 Notably, the reduction of RelA signaling in senescent MCF-7 and A549 cells inhibited the secretion of both TNFα and IFNγ, as well as the surface expression of FAS. These data demonstrate the existence of a RelA-dependent autocrine loop that controls FAS expression by senescent tumor cells. Moreover, these findings suggest a novel anticancer function of the SASP: the sensitization of senescent tumor cells to FAS-mediated apoptosis ().
Figure 1. The induction of senescence promotes FAS upregulation via NFκB as well as the release of chemokines (blue diamonds) that recruit cells of the innate immune system. In addition, senescence results in the secretion of cytokines (red and yellow dots) that act locally within autocrine and paracrine loops, hence sustaining the senescent phenotype and promoting the killing of senescent cells by the immune system.
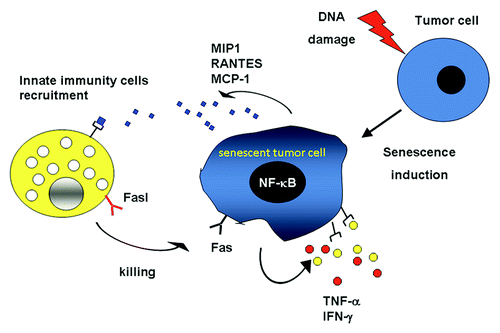
Hence, SASP components form a complicated network that is involved in reinforcing the senescence-associated growth arrest as well as in promoting the recognition and clearance of senescence cells by the immune system. These two biological outcomes of cell senescence are likely to be induced in vivo, in different settings. For instance, senescent melanocytes in nevi are maintained in a growth-arrested state for years, while senescent carcinoma and sarcoma cells are readily cleared by the innate immune system.Citation12,Citation13 It is tempting to speculate that the interplay of distinct transcription factors, including but perhaps not limited to NFκB, C/EBPβ and STAT1 might induce specific SASP components that regulate the outcome of senescence. Further investigation is required to assess how distinct secretory profiles are induced in different physiopathological and experimental settings.
Several lines of evidence point to NFκB as a critical regulator of the SASP. By using in silico approaches, different groups have identified multiple NFκB-controlled genes that are upregulated in senescent cells.Citation21,Citation22 However, the precise role of NFκB in the induction of senescence and in the maintenance of the senescent phenotype is not yet clear, similar to the functional role of NFκB in promoting growth arrest and aging as well as cell growth and cancer. Experimental evidence suggests that NFκB is important for the induction of a growth arrest. For example, Penzo, et al. reported a transient proliferative block in murine fibroblast upon enforced NFκB activation.Citation23 Accordingly, Rovillain and colleagues found that different NFκB target genes are up- or downregulated upon senescent growth arrest, and that interfering with the activation of NFκB bypasses growth arrest.Citation24 Conversely, our data strongly suggest that NFκB does not play a major role in mediating the induction of cell senescence and the associated growth arrest upon DNA damage, as NFκB-depleted cells readily undergo senescence in response to DNA-damaging agents. Rather NFκB seems to be important for the maintenance of the senescent phenotype. In fact, Rovillain, et al. reported that blocking NFκB activity (by the ectopic expression of a superrepressor form of the inhibitory subunit IκBα) overcomes growth arrest and increases the number of growing colonies.Citation24 Accordingly, in our experimental system NFκB-deficient senescent cells escape from the senescence-associated cell cycle arrest (Crescenzi and Leonardi, unpublished data). This effect, however, is seemingly not due to a direct control of cell cycle progression by NFκB, but perhaps to the increased genomic instability observed in the absence of functional NFκB.Citation25,Citation26 In addition, as secreted proteins play a critical role in the maintenance of some forms of OIS,Citation8,Citation10 it is also possible that quantitative and/or qualitative differences in the SASP (for instance, a loss of IL-8 secretion) might account for the increased escape of NFκB-deficient cells from the induction of senescence.
We and others have demonstrated that the secretion of some SASP components depends upon the transcriptional activity of NFκB.Citation15,Citation24,Citation27 Such cytokines and chemokines are largely pro-inflammatory. However, at odds from cancer cells that take advantage of the inflammatory microenvironment and of the recruitment of inflammatory cells, senescent cells are cleared as a consequence of the SASP. Indeed, senescent cells upregulate FAS expression and recruit inflammatory cells to mediate their own clearance. These effects appear to be mediated by NFκB via an autocrine loop requiring TNFα and IFNγ. Interestingly, a similar autocrine loop involving TNFα has also been demonstrated to mediate the killing of cells in response to high doses of etoposide.Citation28 It is possible that NFκB activation is necessary for the establishment of the SASP, and that its functional outcome, that is pro-tumor vs. antitumor, depends upon the microenvironment and/or subtle differences in the cytokine composition of the SASP. Indeed, cells lacking p53 are known to secrete markedly higher levels of most SASP factors, suggesting that one of the ways whereby p53 suppresses tumorigenesis is by modulating the inflammatory microenvironment.Citation5
From this perspective, senescence and NFκB may be considered a Trojan horse that we may harness against cancer with a dual advantage: (1) senescence may be triggered with a relatively low dose of anticancer agents (which results in less severe side effects) and (2) senescent cells—via the SASP—activate the immune system (hence resulting, at least in some scenarios, in improved antitumor effects).
References
- d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003; 426:194 - 8; http://dx.doi.org/10.1038/nature02118; PMID: 14608368
- Suram A, Kaplunov J, Patel PL, Ruan H, Cerutti A, Boccardi V, et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J 2012; 31:2839 - 51; http://dx.doi.org/10.1038/emboj.2012.132; PMID: 22569128
- Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res 1999; 59:3761 - 7; PMID: 10446993
- Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010; 5:99 - 118; http://dx.doi.org/10.1146/annurev-pathol-121808-102144; PMID: 20078217
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008; 6:2853 - 68; http://dx.doi.org/10.1371/journal.pbio.0060301; PMID: 19053174
- Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol 2008; 20:150 - 5; http://dx.doi.org/10.1016/j.ceb.2008.01.007; PMID: 18353625
- Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002; 109:335 - 46; http://dx.doi.org/10.1016/S0092-8674(02)00734-1; PMID: 12015983
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008; 133:1019 - 31; http://dx.doi.org/10.1016/j.cell.2008.03.039; PMID: 18555778
- Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A 2009; 106:17031 - 6; http://dx.doi.org/10.1073/pnas.0905299106; PMID: 19805069
- Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008; 133:1006 - 18; http://dx.doi.org/10.1016/j.cell.2008.03.038; PMID: 18555777
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell 2008; 134:657 - 67; http://dx.doi.org/10.1016/j.cell.2008.06.049; PMID: 18724938
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445:661 - 5; http://dx.doi.org/10.1038/nature05541; PMID: 17251932
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656 - 60; http://dx.doi.org/10.1038/nature05529; PMID: 17251933
- Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009; 113:3503 - 11; http://dx.doi.org/10.1182/blood-2008-08-173914; PMID: 19098271
- Crescenzi E, Pacifico F, Lavorgna A, De Palma R, D’Aiuto E, Palumbo G, et al. NF-κB-dependent cytokine secretion controls Fas expression on chemotherapy-induced premature senescent tumor cells. Oncogene 2011; 30:2707 - 17; http://dx.doi.org/10.1038/onc.2011.1; PMID: 21278794
- Ouaaz F, Li M, Beg AA. A critical role for the RelA subunit of nuclear factor kappaB in regulation of multiple immune-response genes and in Fas-induced cell death. J Exp Med 1999; 189:999 - 1004; http://dx.doi.org/10.1084/jem.189.6.999; PMID: 10075983
- Bretz JD, Arscott PL, Myc A, Baker JR Jr.. Inflammatory cytokine regulation of Fas-mediated apoptosis in thyroid follicular cells. J Biol Chem 1999; 274:25433 - 8; http://dx.doi.org/10.1074/jbc.274.36.25433; PMID: 10464273
- Fluhr H, Krenzer S, Stein GM, Stork B, Deperschmidt M, Wallwiener D, et al. Interferon-gamma and tumor necrosis factor-alpha sensitize primarily resistant human endometrial stromal cells to Fas-mediated apoptosis. J Cell Sci 2007; 120:4126 - 33; http://dx.doi.org/10.1242/jcs.009761; PMID: 18003704
- Kühnel F, Zender L, Paul Y, Tietze MK, Trautwein C, Manns M, et al. NFkappaB mediates apoptosis through transcriptional activation of Fas (CD95) in adenoviral hepatitis. J Biol Chem 2000; 275:6421 - 7; http://dx.doi.org/10.1074/jbc.275.9.6421; PMID: 10692445
- Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 2011; 30:1536 - 48; http://dx.doi.org/10.1038/emboj.2011.69; PMID: 21399611
- Hardy K, Mansfield L, Mackay A, Benvenuti S, Ismail S, Arora P, et al. Transcriptional networks and cellular senescence in human mammary fibroblasts. Mol Biol Cell 2005; 16:943 - 53; http://dx.doi.org/10.1091/mbc.E04-05-0392; PMID: 15574883
- Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev 2007; 21:3244 - 57; http://dx.doi.org/10.1101/gad.1588507; PMID: 18055696
- Penzo M, Massa PE, Olivotto E, Bianchi F, Borzi RM, Hanidu A, et al. Sustained NF-kappaB activation produces a short-term cell proliferation block in conjunction with repressing effectors of cell cycle progression controlled by E2F or FoxM1. J Cell Physiol 2009; 218:215 - 27; http://dx.doi.org/10.1002/jcp.21596; PMID: 18803232
- Rovillain E, Mansfield L, Caetano C, Alvarez-Fernandez M, Caballero OL, Medema RH, et al. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene 2011; 30:2356 - 66; http://dx.doi.org/10.1038/onc.2010.611; PMID: 21242976
- Wang J, Jacob NK, Ladner KJ, Beg A, Perko JD, Tanner SM, et al. RelA/p65 functions to maintain cellular senescence by regulating genomic stability and DNA repair. EMBO Rep 2009; 10:1272 - 8; http://dx.doi.org/10.1038/embor.2009.197; PMID: 19779484
- Gapuzan ME, Schmah O, Pollock AD, Hoffmann A, Gilmore TD. Immortalized fibroblasts from NF-kappaB RelA knockout mice show phenotypic heterogeneity and maintain increased sensitivity to tumor necrosis factor alpha after transformation by v-Ras. Oncogene 2005; 24:6574 - 83; PMID: 16027734
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev 2011; 25:2125 - 36; http://dx.doi.org/10.1101/gad.17276711; PMID: 21979375
- Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell 2011; 145:92 - 103; http://dx.doi.org/10.1016/j.cell.2011.02.023; PMID: 21458669