Abstract
A large number of studies have been published over the last two decades examining molecular mechanisms of antifungal resistance in Candida species. However, few of these studies have explored how such mechanisms influence the host immune response to this opportunistic pathogen. With recent advances in our understanding of host immunity to Candida, a body of emerging literature has begun to explore how intrinsic and adaptive resistance mechanisms in Candida alter host immune system evasion and detection, which could have important implications for understanding (1) why certain resistance mechanisms and Candida species predominate in certain patient populations, (2) the biological context for understanding why high in vitro levels of resistance in may not necessarily correlate with risk of drug failure in vivo and (3) insight into effective immunotherapeutic strategies for combatting Candida resistance. Although this area of research is still in its infancy, two themes are emerging: First, the immunoevasion and intracellular persistence of C. glabrata may be a key factor in the capability of this species to persist in the course of multiple antifungal treatments and develop multidrug resistance. Second, changes in the cell wall associated with antifungal resistance often favor evasion for the host immune response.
Introduction
Candida species are capable of a wide spectrum of infections in human hosts, ranging from benign colonization of the skin and mucosal surfaces to invasion of the bloodstream with dissemination to internal organs. The most common risk factors for invasive candidiasis include major surgery, especially involving the abdomen, immunosuppression (e.g., neutropenia, glucocorticoids and immunomodulators) and many supportive care measures used in the critically ill patient such as broad-spectrum antimicrobials, total parenteral nutrition, renal replacement therapies and central venous catheters.Citation1 The ubiquity of these risk factors explains, in part, the continuing high prevalence of Candida infections in cancer, transplant and ICU patient populations.Citation2,Citation3 Although the prompt administration of effective systemic antifungal therapy can significantly reduce the morbidity and mortality associated with invasive candidiasis, increasing rates of antifungal resistance, particularly among C. glabrata, are threatening to diminish the efficacy of current frontline agents for invasive candidiasis.Citation4-Citation6
A multitude of papers have been published over the last two decades examining the molecular mechanisms of virulence and antifungal resistance in Candida spp. Few of these studies have explored how antifungal resistance mechanisms alter pathogen recognition by the innate immune system, or conversely how host immunological responses shape the evolution antifungal resistance in vivo. Yet a number of recent studies have begun to explore how the microevolution of antifungal resistance in vivo may be shaped by intact or residual host immune responses. Indeed, the host immune response may act as a “second drug” (if not the primary drug) that allows emergence of a resistant subpopulation that gives rise to a breakthrough infection. An improved understanding of the interplay between resistance mechanisms and the host immune response could broaden our understanding of the antifungal resistance landscape in Candida spp and possibly help prioritize drug resistance/pathogen mechanisms that are most likely to emerge in patients. These studies could also aid our understanding why high MICs for some drug-pathogen combinations have limited utility for predicting clinical failure of therapy in patients. In this review, we will examine the emerging data on how antifungal resistance mechanisms alter host immune response to Candida, and project the possible clinical and laboratory implications of these interactions for interpreting susceptibility testing and treating patients with invasive candidiasis.
Overview of Host Immunity to Invasive Candidiasis
Until recently, relatively little was known about how the host immune differentiated benign colonizing yeast forms of Candida from invasive hyphal forms and what triggers were responsible for activation of the inflammatory response. The discovery of Toll-like receptors (TLRs) in the 1990s heralded a revolution in knowledge of innate immunity that revealed a diverse array of receptors and pathways in leukocytes and epithelial cells capable of detecting specific pathogen-associated molecular patterns (PAMPs) expressed at various stages of Candida growth.Citation7-Citation9 Progress since these early discoveries have led to an integrated model for how the host immune response recognizes Candida albicans through pathogen recognition receptors (PRRs) and initiates the early inflammatory response as well as adaptive immunity. A number of excellent reviews have been recently published on this topic.Citation8,Citation10,Citation11 Therefore, the model for host response to Candida is only briefly summarized below.
Morphogenesis and the cell wall
Candida species are capable of growth as yeast, pseudohyphal or hyphal forms. When C. albicans infect humans and animals, hyphae predominate at the primary site of infection in epithelial layers and tissue, whereas yeast forms found on the epithelial cell surface or merging from penetrating hyphae in surrounding tissue ().Citation9,Citation10 The capacity to undergo the reversible yeast-hyphal switch has been shown to be an essential virulence trait of C. albicans.Citation11
Figure 1. Pathogenesis and host immune response to invasive candidiasis. This figure was recreated in a different format from the review by Gow et al.Citation8
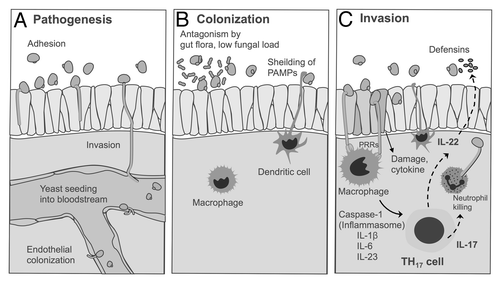
The yeast to hyphal transition is also associated with marked changes in the organization of cell wall carbohydrates and proteins.Citation8 The cell wall of C. albicans is organized into two major layers: an outer layer consisting of glycoproteins (O- and N-linked mannose polymers, mannoproteins) as well as an inner layer containing skeletal polysaccharides (chitin, β-1,3-glucan and β-1,6-glucan).Citation8 For the outer layer, yeast to hyphal transition causes changes in the type of cell surface mannans that are expressed, as well as the highly regulated production of proteins that play a role in adhesion and invasion of epithelial cells.Citation8 For the inner layer, the yeast to hyphal transition has been shown to alter the organization and concentration of structural polysaccharides, including a 3- to 5-fold increase in cell wall chitin and decreased surface exposure of immunogenic β-glucans.Citation8 These changes could be especially important to the host immune response, as the carbohydrates and proteins found in the cell wall represent the major PAMPs used by immune cells for detecting Candida invasion. Hence, the masking of immunostimulatory cell walls glucans and fortification of the hyphal cell wall with less immunogenic chitin could be key immune evasion strategy employed by Candida species during early stages of infection.Citation8
Host immune response to Candida
The first encounter with the host immune response occurs at the epithelium, where the mucosa possesses a complex system for differentiating harmless colonizing yeast from invasive hyphal forms.Citation10,Citation12 Consequently, Candida can colonize but do not typically invade through the mucosal or epithelial layers unless they are damaged resulting in mucosal infection, or in the case of immunocompromised patients, could cause invasive disease that spreads via the bloodstream to distal organs. Inflammatory reactions to yeast forms of Candida are limited in healthy hosts by a low fungal load that is held in check by competing bacterial flora, and cellular morphotype of the fungus, which has limited surface exposure of PAMPs such as β-1,3-D-glucan in the yeast form ().Citation8 As yeast transition to the invasive hyphal morphotype, they exhibit a greater capacity for endocytosis and damage of epithelium, which causes the release of immunogenic cell wall constituents and proinflammatory cytokines and chemokines from the epithelium. Cytokine and chemokine release acts as the initial trigger for attracting monocytes and neutrophils in the circulation, as well as tissue macrophages (). These cells express a repertoire of PAMP receptors including TLRs (i.e., TLR2 and TLR4), which aid in the discrimination of yeast vs. hyphal morphotypes, and C-type lectin receptors (e.g., Dectin-1, Dectin-2, mannose receptor, DC-sign, Mincle and others), which recognize the matrix of glycosylated proteins (mannoproteins) and glucan and chitin polysaccharides that are the major structural elements of the fungal cell wall.Citation8,Citation10
The pattern of PRR ligation that occurs when epithelial or immune cells “taste” Candida cells initiates specific and redundant pathways of immune cell activation leading to cytokine production, phagocytosis, and fungal killing (). This activation pattern also shapes the subsequent pattern of T-cell activation. For example, the balance between TLR2 and TLR4 activation by Candida cell morphotypes determines the dominant type of T helper cell response.Citation11 Ligation of TLR4 is more prominent with the PRRs expressed in yeast form of Candida, which induces a pro-inflammatory response resulting in the production TH1-type cytokines such as INF-γ that boost Candida killing.Citation9 In contrast, hyphal forms of Candida preferentially activate TLR2, which induce a much weaker TH1-response, thus promoting conditions favorable for a TH2 or T-regulatory cell expansion driven by IL-10 that in experimental infections models is associated with reduced Candida clearance.Citation9,Citation13,Citation14 Hence, the shift from the yeast to hyphal morphotype in Candida may represent a key evasive mechanism employed by the pathogen to downregulate protective host immune responses.Citation9
Similar to epithelial cells, tissue macrophages and dendritic cells monitor the microbial flora on the epithelial surface. The mechanism that allows these cells to discriminate between colonizing yeast and invasive hyphal forms was not well understood until the role of inflammasomes and TH17 cells were elucidated for host response to infection and autoimmunity.Citation15 Activation of the PRRs that recognize Candida cell wall β-glucan (Dectin-1), phospholipomannans (TLR2) and mannan (macrophage mannose receptor) induce transcription of pro IL-1β, that under colonizing conditions is not processed to its active form because of limited availability of caspase-1.Citation8,Citation16 However, epithelial damage triggers the activation of the NLRP3 inflammasome in macrophages and dendritic cells, which leads to the activation of caspase-1 and processing of pro IL-1β into the active cytokine form.Citation15 IL-1β subsequently induces TH17-type response, which includes the production of IL-17, IL-22, neutrophil recruitment for hyphal killing, and boosting of epithelial cell responses through production of defensins ().Citation15 Interestingly, a recent report by Zelante et al. suggests that C. albicans can directly sense IL-17A in human hosts, resulting in downregulation of signal transduction pathways, increased adhesion and filamentous growth as well as enhanced biofilm formation that facilitates resistance to attack by phagocytic cells.Citation17
While this model for host immunity to Candida is far from complete, it suggests that the host response is highly adapted and evolved to detect changes in the fungal cell wall and morphology. Growth of fungal cells under varying culture conditions can markedly change cell wall content even if cell morphology is unaltered.Citation8 Therefore, it seems likely that antifungal therapy in vivo and compensatory resistance mechanisms influencing cell wall composition and organization would similarly alter detection by the host immune cells. The nature of these interactions may have important consequences on the emergence of resistance in vivo and its impact on the host.
Intrinsically Resistant Candida Species
Approximately 95% of all invasive Candida infections are caused by five species: C. albicans, C. glabrata, C. parapsilosis, C. tropicalis and C. krusei.Citation18 Among these species, only C. glabrata and C. krusei are numerically increasing in some geographic areas such as the United States due in part to their intrinsic and acquired resistance to azoles and other commonly used antifungal agents.Citation18 The prevalence of C. glabrata infections is highest among severely ill patients greater than 60 y of age, with one-third of bloodstream infections caused by this species.Citation19 Importantly, reports of bloodstream infections due to C. glabrata resistant to multiple triazoles and echinocandins have increased in recent years.Citation6 In a review of data from population-based and lab-based surveillance programs in the US, Pfaller and colleagues noted that 9.7% of C. glabrata strains were resistant to fluconazole, of which 99% were cross-resistant to voriconazole and 8–9% were also cross-resistant to anidulafungin, caspofungin and micafungin.Citation6 In contrast, no echinocandin-resistant strains were detected in isolates collected from 2001–2004. These data suggest that while a majority of C. glabrata remains susceptible to echinocandin antifungals, the recent increase in MDR strains may represent an ominous trend justifying continued surveillance and antimicrobial susceptibility testing.
The haploid nature of C. glabrata genome makes the pathogen particularly well suited for acquiring and expressing MDR resistance traits in the presence of drug pressure.Citation20,Citation21 However, C. glabrata lacks several key virulence factors reported to be essential for virulence in diploid organisms C. albicans.Citation21 For example, C. glabrata is the only Candida species that does not form pseudohyphae at temperatures above 37°C or secrete hydrolases that have been shown to be essential for tissue invasion and persistence in C. albicans.Citation22 C. glabrata also behaves differently from other Candida species in the classic mouse intravenous infection challenge.Citation23 Whereas C. albicans and C. tropicalis induce severe systemic inflammation and rapidly progressive invasion of kidneys and brain, depending on the mouse strain and inoculum level, inoculation with C. krusei and C. parapsilosis are not lethal even at high inoculum levels, and are eventually cleared by infected animals. On the other hand, intravenous challenge with even high inoculum of C. glabrata is non-lethal in immunocompentent mice, but produces a sustained high fungal burden in animal kidneys that appears to be tolerated by animals with minimal inflammation.Citation23 Remarkably, C. glabrata can be isolated from infected immunocompetent mice over several weeks, without evidence of rapid immune system clearance observed with other Candida species.Citation24 These observations suggest that C. glabrata may have a fundamentally greater capacity for immune evasion, which could favor its persistence and emergence as a MDR pathogen in the setting of continuous or repetitive pressure with antifungal agents.
The concept that immune evasion is a key element in C. glabrata infection, and by extension the emergence of multidrug resistance, is also supported by studies that have examined interactions of this species with phagocytic cells.Citation25 C. glabrata can survive attack by phagocytes and even replicate inside macrophages after engulfment.Citation25 Sieder and colleagues recently demonstrated that intracellularly proliferating C. glabrata in human macrophages do not elicit the production of reactive oxygen species and only marginally induce production of pro- or anti-inflammatory cytokines.Citation26 Interestingly, phagosomes containing viable C. glabrata, but not heat-killed yeast, failed to recruit cathepsin D and were only weakly acidified. Therefore it appeared that viable C. glabrata was able to subvert normal macrophage phagosome maturation, survive and replicate within these immune cells for considerable periods of time without damaging the host cell or eliciting a proinflammatory immune response.Citation26 While this interaction could be mutually beneficial during commensal carriage, it would be detrimental for clearance of an invasive infection in a debilitated host during invasive infections.Citation26
The exploitation of an intracellular niche as part of an immune evasion and persistence strategy could also favor the development of antifungal resistance in C. glabrata and possibly C. parapsilosis. In a series of recent studies, Baltch et al. and Bopp et al. examined intracellular yeast killing kinetics of voriconazole and echinocandins (caspofungin and micafungin) in macrophages infected with various Candida species.Citation27-Citation30 In contrast to C. krusei where viable CFU counts inside macrophages decreased even in the absence of antifungal exposure, reductions in viable colony forming unit counts of intracellular C. parapsilosis and C. glabrata required voriconazole or micafungin concentrations that were often 2.5 to 5 times higher than the extracellular MIC.Citation28 Intracellular anti-candidal activity could be improved if these agents where administered in combination, or in macrophages primed with granulocyte-macrophage colony stimulating factor.
Collectively, these studies provide a plausible hypothesis of how C. glabrata may be capable of surviving and persisting in the face of prolonged antifungal therapy to later emerge in a weakened host. Treatment strategies that improve the intracellular activity of antifungals or host immune responses may be a key pathway for reducing antifungal resistance of treating persistent C. glabrata fungemia.
Amphotericin B acquired resistance
Amphotericin B exhibits fungicidal effects in Candida species by binding to ergosterol in the fungal cell membrane forming pores that cause membrane destabilization and leakage of intracellular contents. Amphotericin B resistance in Candida is generally assumed to be rare, although many broth-based methods used for detecting resistance in Candida species may lack the sensitivity to reliably detect resistance in vitro.Citation4 Candida species for which MICs > 1 mg/L are unusual, but at the very least, may require higher doses of amphotericin B for optimal treatment.Citation4,Citation31 Compared with C. albicans, C. glabrata and C. krusei are less susceptible to amphotericin B in vitro and display delayed killing kinetics by time-kill studies.Citation4 C. lusitaniae is notorious for developing resistance during amphotericin B therapy, although the species is often susceptible upon initial isolation from the bloodstream.Citation32 The acquired resistance in this species has been linked to high-frequency phenotypic switching from susceptibility to resistance upon amphotericin B exposure.Citation33,Citation34 To our knowledge no study has explored differences in host immune responses or virulence for the amphotericin B susceptible vs. resistant phenotypes of C. lusitaniae.
The most commonly cited mechanisms of amphotericin B resistance in Candida species include alterations in ergosterol biosynthesis leading to a decrease in the amount of ergosterol in the plasma membrane or increased production of catalases reducing drug-associated oxidative damage.Citation32 However, amphotericin B-resistant strains are rarely isolated from patients suggesting that either the sterol substitutions may be associated with significant fitness costs to infecting isolates in vivo, or possibly enhanced eradication by the host immune response. Indeed, inactivation of enzyme sterol Δ5,6-desaturase (ERG3) in C. albicans, which results in ergsoterol sterol membrane substitutions and diminished fluconazole and amphotericin B susceptibility, produces C. albicans strains locked in the yeast form with attenuated virulence in animal models.Citation35 A recent report from Vale-Silva and colleagues, however, reported that unlike C. albicans, loss-of-function Δ5,6-desaturase (ERG3) mutations in C. glabrata do not necessarily result in decreased virulence in animal models.Citation36
Triazole acquired resistance
Triazoles inhibit 14-α-demethylase, an enzyme responsible for conversion of lanosterol to ergosterol in pathogenic fungi. Inhibition of this rate-limiting step in the ergosterol biosynthesis pathway results in abnormal cell membrane fluidity and function, arresting fungal cell growth. Several resistance mechanisms are commonly associated with triazole resistance in Candida species. First, mutations in the gene encoding the drug target Erg11 alter the drug-binding domain of triazoles, reducing the potency of some, but not necessarily all triazoles.Citation32,Citation37 Second, high level triazole resistance may result from overexpression of genes involved in the sterol biosynthesis pathway as well as upregulation of two families of efflux pumps, the ATP-binding cassette (ABC) (Cdr1 and Cdr2) and the major facilitator superfamily (MFS) Mdr1.Citation38 Frequently, the co-expression of these resistance mechanisms results in cross-resistance to all triazoles, isolated most frequently in patients with breakthrough C. glabrata fungemia.Citation32 A less common mechanism of resistance in C. glabrata involves mitochondrial dysfunction in C. glabrata, resulting in the triazole-resistant “petite mutant” growth phenotype in vitro that is resistant to triazoles.Citation39,Citation40
Biofilm formation is clearly an important virulence trait and resistance mechanism for chronic or relapsing Candida infections that acts as a physical barrier protecting underlying cells from phagocytes and limiting drug penetration.Citation41-Citation43 Recent evidence suggest that β-1,3-glucans are a major component of Candida biofilms and may directly bind triazole antifungals such as fluconazole.Citation41,Citation43 Because drug efflux pumps are upregulated when cells grow in biofilm condition,Citation44 it is possible that selection of mutants overexpressing efflux mechanisms during drug therapy may favor biofilm-oriented growth and escape from the host immune system.Citation42,Citation45,Citation46
Several studies have surveyed the impact of these acquired triazole resistance mechanisms on virulence in Candida species (). Graybill and colleagues examined this question in C. albicans by testing the virulence of azole-resistant isolates in a mouse model of invasive candidiasis compared with their azole-susceptible susceptible parental isolate.Citation47 The authors concluded that no direct relationship between fluconazole susceptibility and survival (virulence) was evident. A similar study by Schulz et al. using clonally related C. albicans strains from patients with oropharyngeal candidiasis reported that while the fluconazole-susceptible strain was more virulent and exhibited faster growth kinetics and increased biofilm formation, the resistant strain adhered more avidly to epithelial cells facilitating colonization.Citation48
Table 1. Acquired Candida resistance mechanisms and potential impact on host pathogen interaction
The impact of antifungal resistance on the host immune response and pathogen virulence may differ, however, for C. glabrata. Ferrari and colleagues recently reported that gain of function mutations in the transcriptional regulator CgPDR1—the key modulator of Cdr1 and Cdr2 expression in C. glabrata, was associated not only with higher levels of in vitro/in vivo resistance to fluconazole, but also increased virulence and “dominance” of the fungal population in mice even in the absence of drug selection.Citation49,Citation50 Enhanced in vivo virulence has also been reported in other fluconazole-resistant C. glabrata isolates selected in vivo where triazole resistance developed from mitochondrial DNA deficiency independent of gain-of-function mutations in drug efflux pumps.Citation49 Interestingly, these petite mutants displayed enhanced expression of stress response pathways and cell wall remodeling, similar to that reported after exposure to echinocandin antifungals.Citation51 Although the specific role of these resistance mechanisms has not yet been fully elucidated with respect to host pathogen interactions, overexpression of drug efflux transporters in other yeast species such as Cryptococcus neoformans has been shown to interfere with lysosome acidification in macrophages to increase intracellular fungal survival.Citation52
Finally, Takahashi and colleagues reported that acquired triazole and echinocandin resistance in C. glabrata was associated with significant changes in the antigenic cell wall mannoprotein structure of C. glabrata.Citation53 Specifically, C. glabrata isolates exhibiting resistance to both itraconazole and micafungin contained very low cell concentrations of β-1,2-linked mannose residues relative to susceptible strains. These β-1,2-linked mannose residues have been previously shown to induce TNF-α synthesis through TLR2.Citation54 Although it was not specifically tested in this study, the authors’ findings would suggest that the echinocandin and triazole-resistant C. glabrata strains would not elicit as potent response (i.e., TNF-α) release from epithelial or phagocytic cells.Citation54
Collectively, these studies suggest that the acquired triazole resistance, particularly in C. glabrata, may be associated with significant and possibly advantageous changes in the fungal cell wall and compensatory mechanisms for evading or surviving the initial host immune response. The acquired host immune evasion mechanisms could further facilitate the capability of this species to persist in the setting of antifungal therapy and mutate into multidrug resistant forms.
Echinocandin acquired resistance
Echinocandin antifungals (anidulafungin, caspofungin and micafungin) are among the most widely prescribed antifungals in patients with invasive candidiasis. Despite some pharmacokinetic differences, all three echinocandins act by inhibiting 1,3-β-D-glucan synthase, thereby disrupting glucan biosynthesis in the cell wall.Citation55 Candida cells exposed to echinocandin concentrations near the MIC have defective cell walls that render the cell susceptible to osmotic lysis. However, even subinhibitory concentrations of echinocandins affect cell wall organization resulting in increased cell surface exposure of immunogenic β-glucans normally hidden by surface mannoproteins, resulting in strong stimulation of immune responses and increases levels of cytokines such as tumor necrosis factor α, interleukin-6 (IL-6), IL-10 and γ-interferon.Citation56 This β-glucan “unmasking” effect has been shown to be sufficient for fungicidal activity in animals with intact innate immunity.Citation57 Interestingly, this “unmaking” effect of β-glucan may persist even in some echinocandin-resistant strains, which may be an important limiting factor for the emergence of some resistant subpopulations during treatment.Citation58,Citation59
Echinocandin resistance in Candida species is most frequently caused by mutations in the genes encoding 1,3-β-D-glucan synthase complex, often in conserved “hot spot” regions of the FKS1 catalytic subunit, although mutations in FKS2 and FKS3 catalytic subunits have also been observed in C. glabrata.Citation60 Mutations in the FKS catalytic subunits alter the kinetics of the glucan synthase enzyme complex resulting in higher inhibitory constant 50% (IC50) and a 50- to several thousand-fold increased kinetic inhibition (ki) for the mutant enzymes for all three echinocandins compared with sensitive wild-type strains.Citation61 Therefore, mutations in the FKS catalytic sites generally result in cross-resistance to all three echinocandins.Citation62
A second pattern of resistance or tolerance to echinocandin fungicidal effects is also sometimes observed when Candida cells are exposed to echinocandin concentrations above the MIC.Citation63 This paradoxical persistence at higher echinocandin concentrations appears to be mediated through fungal cellular homeostatic cell wall remodeling pathways.Citation64 Following echinocandin exposure in Candida albicans, HOG1, CEK1, PKC MAP kinase and Ca2+-calcineurin signaling pathways are upregulated resulting in increased cellular glucan and chitin synthesis, changes in cellular protein, and increased tolerance to echinocandins with paradoxical growth at supra-MIC concentrations.Citation65-Citation67 Interestingly, isolates exposed to paradoxical growth inducing concentrations of glucan synthesis inhibitors such as caspofungin often display reduced ability to activate RAW 264.7 macrophages through Dectin-1 (β-D-glucan) dependent mechanisms (Lewis et al., submitted). Deletion of genes involved in these homeostatic pathways or pharmacologic inhibition (i.e., with calcineurin inhibitors) often reverses paradoxical growth at high echinocandin concentrations and enhances the anti-Candida potency of echinocandins.Citation68,Citation69 Interestingly, the phenomena of echinocandin paradoxical growth varies between the echinocandin tested and Candida species, with paradoxical effects observed most frequently when caspofungin is tested in vitro against in clinical isolates of C. parapsilosis, C. albicans, C. dubliniensis, C. tropicalis and occasionally C. krusei.Citation70 Paradoxical growth was not observed when C. glabrata is treated with echinocandins. Because paradoxical growth of Candida exposed to high echinocandin concentrations is difficult to detect in vivo,Citation71-Citation73 some experts have suggested that this phenomena is only an artifact of in vitro testing. Nevertheless, paradoxical growth clearly represents an adaptive response of the fungus to drug and probably influences host immune responses.
Early studies performed by Douglas and Kurtz examining the effects of FKS1 mutations on echinocandin susceptibility suggested that some mutations associated with high-level echinocandin-resistance may be associated with significant growth defects, impaired yeast to hyphal transition in C. albicans and decreased virulence in vivo.Citation74 Consistent with this observation, a clinical study examining echinocandin-resistant breakthrough C. tropicalis infections in leukemic patients found that resistant isolates rarely caused metastatic infection or sepsis.Citation75 However, generalizations regarding the clinical outcome of echinocandin-resistant strains are difficult, given the heterogeneity and diversity of clinical risk factors of patients in published clinical reports.
Ben-Ami and colleagues recently examined the fitness and virulence costs of FKS1 hot spot mutations associated with echinocandin-resistance in invertebrate and vertebrate models of invasive candidiasis.Citation58 Compared with wild-type stains, C. albicans strains with homozygous FKS1 mutations had reduced catalytic activity of glucan synthase, thicker cell walls attributable to increased cell wall chitin and reduced growth rate and capacity for filamentation.Citation59 FKS1 mutants were hypovirulent in fly and mouse infection models, including mixed growth competition assays with wild-type strains. In vivo virulence was highly correlated with the cell wall chitin content of the infecting strain. Importantly, FKS1 mutants with increased cell wall chitin content induced weaker Dectin-1 dependent inflammatory responses when coincubated with RAW264.7 macrophages compared with Wt or FKS mutant strains that had minimal increases in cell wall chitin. Data concerning the effect of FKS1 mutations on biofilm formation capacity in echinocandin-resistant isolates was less conclusive, but several studies have suggested that FKS1 mutant strains produce biofilm with a less dense matrix but with similar mass as wild-type cells.
These studies were corroborated by investigators from Aberdeen, who reported that C. albicans strains harboring FKS1 mutations often display higher basal production of chitin.Citation64,Citation76 Animals infected with C. albicans cells with elevated cell wall chitin concentrations were similarly resistant to echinocandin treatment.Citation76 Similar to Ben-Ami et al., the mean survival time of mice infected with high-chitin cells was considerably longer than that of mice infected with normal-chitin cells. Interestingly, compensatory increases in cell wall chitin synthesis were also found to be a good indicator if which stain display paradoxical growth at high echinocandin concentrations.Citation65 The investigators were able to demonstrate that chitin purified from Candida albicans cell wall blocked Dectin-1 mediated recognition of human peripheral blood mononuclear cells and murine macrophages, leading to significant reductions in cytokine production.Citation76 Hence, chitin may be a signature of “less invasive” form of the pathogen, and consequently does not invoke as vigorous of immune response as β-D-glucan.
Taken as a whole, these studies demonstrated that C. albicans remodels its cell wall in response to echinocandin therapy and this adaptation can have a significant impact on pathogen virulence and recognition by host immune cells.Citation77 These studies may also provide some explanation of why the prevalence of FKS1 hot spot mutations continues to be low in C. albicans and why some patients with bloodstream infection have limited evidence of visceral dissemination/sepsis, or sometimes paradoxically improved clinical response when infected with echinocandin-resistant strains. Nevertheless, other investigators have reported increased virulence in C. albicans harboring FKS1 mutations,Citation78,Citation79 although immunological responses against these strains were not investigated. The in vivo impact of FKS1 mutations may be very different in C. glabrata vs. C. albicans, as suggested in current epidemiological trends of resistance. These questions will provide fascinating avenues for future laboratory, clinical and epidemiological research of invasive candidiasis.
Possible Clinical Implications
Knowledge of how drug resistance mechanisms affect host immune responses is fundamental to understanding the clinical impact of antifungal resistance in opportunistic pathogens such as C. albicans. As the key elements in the host immune response to Candida become clearer, an evaluation of how these central elements are affected by drug resistance should be a research priority. These studies could shed light on: (1) the potential for resistance spreading in given patient populations, (2) biological context for understanding why high levels of resistance in vitro may not necessarily correlate with high risk of drug failure in vivo or (3) effective immunotherapeutic strategies for combatting resistance. These studies may also help explain why despite developing resistance mechanisms that may favor the spread to different patient populations, many less common Candida species never spread beyond their “classic” host niches.Citation18
Host immunity to Candida spp is complex by design, as are the pathogen’s responses strategies for coexisting with, or escaping the host immune response. This complexity should not prevent the search for patterns of altered host immune responses and pathogen virulence that develop with antifungal resistance (). Such studies will undoubtedly lead to more questions than answers, but the answers are potentially clinically important and may help solve the even larger mystery of “clinical resistance”—the catch-all term for why patients fail antifungal therapy when the pathogen appears susceptible in the laboratory.
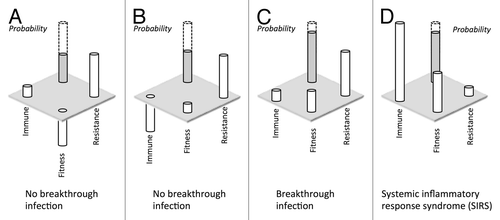
Figure 2. Illustrative hypothetical scenarios for the risk of breakthrough infection with resistant Candida species. In (A), expression of a new resistance mechanisms in the presence of antifungal therapy does not alter host immune detection/elimination, but diminishes pathogen fitness. Therefore, the probability of breakthrough infection is low at the current clinical status of the host. In (B), the expression of the resistance mechanism is not associated with a significant fitness cost but does impact immune evasion strategies, therefore the emergence of the resistant subpopulation in held in check by the immune response. In (C), expression of the resistance mechanism is not associated with significant costs in terms of pathogen fitness or host immune evasion, therefore the resistant subpopulation “emerges” in the presence of antifungal therapy. In (D), the isolate does not express resistance mechanisms in the presence of drug, but the high virulence and strong induction of immune responses lead to sepsis (SIRS). The concept for this figure was derived from review by James Anderson.Citation80
References
- Pappas PG, Kauffman CA, Andes D, Benjamin DK Jr., Calandra TF, Edwards JE Jr., et al, Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503 - 35; http://dx.doi.org/10.1086/596757; PMID: 19191635
- Pappas PG, Alexander BD, Andes DR, Hadley S, Kauffman CA, Freifeld A, et al. Invasive fungal infections among organ transplant recipients: results of the Transplant-Associated Infection Surveillance Network (TRANSNET). Clin Infect Dis 2010; 50:1101 - 11; http://dx.doi.org/10.1086/651262; PMID: 20218876
- Horn DL, Neofytos D, Anaissie EJ, Fishman JA, Steinbach WJ, Olyaei AJ, et al. Epidemiology and outcomes of candidemia in 2019 patients: data from the prospective antifungal therapy alliance registry. Clin Infect Dis 2009; 48:1695 - 703; http://dx.doi.org/10.1086/599039; PMID: 19441981
- Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 2007; 20:133 - 63; http://dx.doi.org/10.1128/CMR.00029-06; PMID: 17223626
- Pfaller MA, Diekema DJ, Gibbs DL, Newell VA, Ellis D, Tullio V, et al, and the Global Antifungal Surveillance Group. Results from the ARTEMIS DISK Global Antifungal Surveillance Study, 1997 to 2007: a 10.5-year analysis of susceptibilities of Candida Species to fluconazole and voriconazole as determined by CLSI standardized disk diffusion. J Clin Microbiol 2010; 48:1366 - 77; http://dx.doi.org/10.1128/JCM.02117-09; PMID: 20164282
- Pfaller MA, Castanheira M, Lockhart SR, Ahlquist AM, Messer SA, Jones RN. Frequency of decreased susceptibility and resistance to echinocandins among fluconazole-resistant bloodstream isolates of Candida glabrata. J Clin Microbiol 2012; 50:1199 - 203; http://dx.doi.org/10.1128/JCM.06112-11; PMID: 22278842
- Netea MG, van der Graaf C, Van der Meer JWM, Kullberg BJ. Toll-like receptors and the host defense against microbial pathogens: bringing specificity to the innate-immune system. J Leukoc Biol 2004; 75:749 - 55; http://dx.doi.org/10.1189/jlb.1103543; PMID: 15075354
- Gow NA, van de Veerdonk FL, Brown AJ, Netea MG. Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nat Rev Microbiol 2012; 10:112 - 22; http://dx.doi.org/10.1038/nrmicro2711; PMID: 22158429
- Netea MG, Brown GD, Kullberg BJ, Gow NA. An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol 2008; 6:67 - 78; http://dx.doi.org/10.1038/nrmicro1815; PMID: 18079743
- Netea MG, Kullberg BJ. Epithelial sensing of fungal invasion. Cell Host Microbe 2010; 8:219 - 20; http://dx.doi.org/10.1016/j.chom.2010.08.008; PMID: 20833371
- Romani L. Immunity to fungal infections. Nat Rev Immunol 2011; 11:275 - 88; http://dx.doi.org/10.1038/nri2939; PMID: 21394104
- Netea MG, Maródi L. Innate immune mechanisms for recognition and uptake of Candida species. Trends Immunol 2010; 31:346 - 53; http://dx.doi.org/10.1016/j.it.2010.06.007; PMID: 20705510
- Netea MG, Van Der Graaf CA, Vonk AG, Verschueren I, Van Der Meer JW, Kullberg BJ. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J Infect Dis 2002; 185:1483 - 9; http://dx.doi.org/10.1086/340511; PMID: 11992285
- Netea MG, Sutmuller R, Hermann C, Van der Graaf CA, Van der Meer JW, van Krieken JH, et al. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J Immunol 2004; 172:3712 - 8; PMID: 15004175
- Cheng SC, van de Veerdonk FL, Lenardon M, Stoffels M, Plantinga T, Smeekens S, et al. The dectin-1/inflammasome pathway is responsible for the induction of protective T-helper 17 responses that discriminate between yeasts and hyphae of Candida albicans. J Leukoc Biol 2011; 90:357 - 66; http://dx.doi.org/10.1189/jlb.1210702; PMID: 21531876
- van de Veerdonk FL, Joosten LA, Shaw PJ, Smeekens SP, Malireddi RK, van der Meer JW, et al. The inflammasome drives protective Th1 and Th17 cellular responses in disseminated candidiasis. Eur J Immunol 2011; 41:2260 - 8; http://dx.doi.org/10.1002/eji.201041226; PMID: 21681738
- Zelante T, Iannitti RG, De Luca A, Arroyo J, Blanco N, Servillo G, et al. Sensing of mammalian IL-17A regulates fungal adaptation and virulence. Nat Commun 2012; 3:683; http://dx.doi.org/10.1038/ncomms1685; PMID: 22353714
- Pfaller MA, Diekema DJ. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol 2010; 36:1 - 53; http://dx.doi.org/10.3109/10408410903241444; PMID: 20088682
- Pfaller MA, Castanheira M, Messer SA, Moet GJ, Jones RN. Variation in Candida spp. distribution and antifungal resistance rates among bloodstream infection isolates by patient age: report from the SENTRY Antimicrobial Surveillance Program (2008-2009). Diagn Microbiol Infect Dis 2010; 68:278 - 83; http://dx.doi.org/10.1016/j.diagmicrobio.2010.06.015; PMID: 20846808
- Chapeland-Leclerc F, Hennequin C, Papon N, Noël T, Girard A, Socié G, et al. Acquisition of flucytosine, azole, and caspofungin resistance in Candida glabrata bloodstream isolates serially obtained from a hematopoietic stem cell transplant recipient. Antimicrob Agents Chemother 2010; 54:1360 - 2; http://dx.doi.org/10.1128/AAC.01138-09; PMID: 20038613
- Fidel PL Jr., Vazquez JA, Sobel JD. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev 1999; 12:80 - 96; PMID: 9880475
- Kaur R, Domergue R, Zupancic ML, Cormack BP. A yeast by any other name: Candida glabrata and its interaction with the host. Curr Opin Microbiol 2005; 8:378 - 84; http://dx.doi.org/10.1016/j.mib.2005.06.012; PMID: 15996895
- Maccallum DM. Hosting infection: experimental models to assay Candida virulence. Int J Microbiol 2012; 2012:363764; http://dx.doi.org/10.1155/2012/363764; PMID: 22235206
- Jacobsen ID, Brunke S, Seider K, Schwarzmüller T, Firon A, d’Enfért C, et al. Candida glabrata persistence in mice does not depend on host immunosuppression and is unaffected by fungal amino acid auxotrophy. Infect Immun 2010; 78:1066 - 77; http://dx.doi.org/10.1128/IAI.01244-09; PMID: 20008535
- Seider K, Heyken A, Lüttich A, Miramón P, Hube B. Interaction of pathogenic yeasts with phagocytes: survival, persistence and escape. Curr Opin Microbiol 2010; 13:392 - 400; http://dx.doi.org/10.1016/j.mib.2010.05.001; PMID: 20627672
- Seider K, Brunke S, Schild L, Jablonowski N, Wilson D, Majer O, et al. The facultative intracellular pathogen Candida glabrata subverts macrophage cytokine production and phagolysosome maturation. J Immunol 2011; 187:3072 - 86; http://dx.doi.org/10.4049/jimmunol.1003730; PMID: 21849684
- Baltch AL, Lawrence DA, Ritz WJ, Andersen NJ, Bopp LH, Michelsen PB, et al. Effects of echinocandins on cytokine/chemokine production by human monocytes activated by infection with Candida glabrata or by lipopolysaccharide. Diagn Microbiol Infect Dis 2012; 72:226 - 33; http://dx.doi.org/10.1016/j.diagmicrobio.2011.11.004; PMID: 22209510
- Baltch AL, Bopp LH, Smith RP, Ritz WJ, Michelsen PB. Anticandidal effects of voriconazole and caspofungin, singly and in combination, against Candida glabrata, extracellularly and intracellularly in granulocyte-macrophage colony stimulating factor (GM-CSF)-activated human monocytes. J Antimicrob Chemother 2008; 62:1285 - 90; http://dx.doi.org/10.1093/jac/dkn361; PMID: 18772160
- Bopp LH, Baltch AL, Ritz WJ, Michelsen PB, Smith RP. Antifungal effect of voriconazole on intracellular Candida glabrata, Candida krusei and Candida parapsilosis in human monocyte-derived macrophages. J Med Microbiol 2006; 55:865 - 70; http://dx.doi.org/10.1099/jmm.0.46393-0; PMID: 16772413
- Baltch AL, Bopp LH, Smith RP, Ritz WJ, Carlyn CJ, Michelsen PB. Effects of voriconazole, granulocyte-macrophage colony-stimulating factor, and interferon gamma on intracellular fluconazole-resistant Candida glabrata and Candida krusei in human monocyte-derived macrophages. Diagn Microbiol Infect Dis 2005; 52:299 - 304; http://dx.doi.org/10.1016/j.diagmicrobio.2005.02.017; PMID: 15893901
- Pfaller MA, Espinel-Ingroff A, Canton E, Castanheira M, Cuenca-Estrella M, Diekema DJ, et al. Wild-Type MIC Distributions and Epidemiological Cutoff Values for Amphotericin B, Flucytosine, and Itraconazole and Candida spp. as Determined by CLSI Broth Microdilution. J Clin Microbiol 2012; 50:2040 - 6; http://dx.doi.org/10.1128/JCM.00248-12; PMID: 22461672
- Pfaller MA. Antifungal drug resistance: mechanisms, epidemiology, and consequences for treatment. Am J Med 2012; 125:Suppl S3 - 13; http://dx.doi.org/10.1016/j.amjmed.2011.11.001; PMID: 22196207
- Chen SC, Marriott D, Playford EG, Nguyen Q, Ellis D, Meyer W, et al, Australian Candidaemia Study. Candidaemia with uncommon Candida species: predisposing factors, outcome, antifungal susceptibility, and implications for management. Clin Microbiol Infect 2009; 15:662 - 9; http://dx.doi.org/10.1111/j.1469-0691.2009.02821.x; PMID: 19614718
- Atkinson BJ, Lewis RE, Kontoyiannis DP. Candida lusitaniae fungemia in cancer patients: risk factors for amphotericin B failure and outcome. Med Mycol 2008; 46:541 - 6; http://dx.doi.org/10.1080/13693780801968571; PMID: 19180749
- Chau AS, Gurnani M, Hawkinson R, Laverdiere M, Cacciapuoti A, McNicholas PM. Inactivation of sterol Delta5,6-desaturase attenuates virulence in Candida albicans. Antimicrob Agents Chemother 2005; 49:3646 - 51; http://dx.doi.org/10.1128/AAC.49.9.3646-3651.2005; PMID: 16127034
- Vale-Silva LA, Coste AT, Ischer F, Parker JE, Kelly SL, Pinto E, et al. Azole resistance by loss of function of the sterol Δ⁵,⁶-desaturase gene (ERG3) in Candida albicans does not necessarily decrease virulence. Antimicrob Agents Chemother 2012; 56:1960 - 8; http://dx.doi.org/10.1128/AAC.05720-11; PMID: 22252807
- Perlin DS. Antifungal drug resistance: do molecular methods provide a way forward?. Curr Opin Infect Dis 2009; 22:568 - 73; http://dx.doi.org/10.1097/QCO.0b013e3283321ce5; PMID: 19741524
- MacCallum DM, Coste A, Ischer F, Jacobsen MD, Odds FC, Sanglard D. Genetic dissection of azole resistance mechanisms in Candida albicans and their validation in a mouse model of disseminated infection. Antimicrob Agents Chemother 2010; 54:1476 - 83; http://dx.doi.org/10.1128/AAC.01645-09; PMID: 20086148
- Bouchara JP, Zouhair R, Le Boudouil S, Renier G, Filmon R, Chabasse D, et al. In-vivo selection of an azole-resistant petite mutant of Candida glabrata. J Med Microbiol 2000; 49:977 - 84; PMID: 11073151
- Sanglard D, Ischer F, Bille J. Role of ATP-binding-cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob Agents Chemother 2001; 45:1174 - 83; http://dx.doi.org/10.1128/AAC.45.4.1174-1183.2001; PMID: 11257032
- Silva S, Henriques M, Martins A, Oliveira R, Williams D, Azeredo J. Biofilms of non-Candida albicans Candida species: quantification, structure and matrix composition. Med Mycol 2009; 47:681 - 9; http://dx.doi.org/10.3109/13693780802549594; PMID: 19888800
- Nett JE, Crawford K, Marchillo K, Andes DR. Role of Fks1p and matrix glucan in Candida albicans biofilm resistance to an echinocandin, pyrimidine, and polyene. Antimicrob Agents Chemother 2010; 54:3505 - 8; http://dx.doi.org/10.1128/AAC.00227-10; PMID: 20516280
- Nett J, Lincoln L, Marchillo K, Massey R, Holoyda K, Hoff B, et al. Putative role of beta-1,3 glucans in Candida albicans biofilm resistance. Antimicrob Agents Chemother 2007; 51:510 - 20; http://dx.doi.org/10.1128/AAC.01056-06; PMID: 17130296
- Ramage G, Bachmann S, Patterson TF, Wickes BL, López-Ribot JL. Investigation of multidrug efflux pumps in relation to fluconazole resistance in Candida albicans biofilms. J Antimicrob Chemother 2002; 49:973 - 80; http://dx.doi.org/10.1093/jac/dkf049; PMID: 12039889
- Nobile CJ, Fox EP, Nett JE, Sorrells TR, Mitrovich QM, Hernday AD, et al. A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell 2012; 148:126 - 38; http://dx.doi.org/10.1016/j.cell.2011.10.048; PMID: 22265407
- Robbins N, Uppuluri P, Nett J, Rajendran R, Ramage G, Lopez-Ribot JL, et al. Hsp90 governs dispersion and drug resistance of fungal biofilms. PLoS Pathog 2011; 7:e1002257; http://dx.doi.org/10.1371/journal.ppat.1002257; PMID: 21931556
- Graybill JR, Montalbo E, Kirkpatrick WR, Luther MF, Revankar SG, Patterson TF. Fluconazole versus Candida albicans: a complex relationship. Antimicrob Agents Chemother 1998; 42:2938 - 42; PMID: 9797229
- Schulz B, Weber K, Schmidt A, Borg-von Zepelin M, Ruhnke M. Difference in virulence between fluconazole-susceptible and fluconazole-resistant Candida albicans in a mouse model. Mycoses 2011; 54:e522 - 30; http://dx.doi.org/10.1111/j.1439-0507.2010.01970.x; PMID: 21605180
- Ferrari S, Sanguinetti M, De Bernardis F, Torelli R, Posteraro B, Vandeputte P, et al. Loss of mitochondrial functions associated with azole resistance in Candida glabrata results in enhanced virulence in mice. Antimicrob Agents Chemother 2011; 55:1852 - 60; http://dx.doi.org/10.1128/AAC.01271-10; PMID: 21321146
- Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, et al. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog 2009; 5:e1000268; http://dx.doi.org/10.1371/journal.ppat.1000268; PMID: 19148266
- Liu TT, Lee RE, Barker KS, Lee RE, Wei L, Homayouni R, et al. Genome-wide expression profiling of the response to azole, polyene, echinocandin, and pyrimidine antifungal agents in Candida albicans. Antimicrob Agents Chemother 2005; 49:2226 - 36; http://dx.doi.org/10.1128/AAC.49.6.2226-2236.2005; PMID: 15917516
- Sanguinetti M, Posteraro B, La Sorda M, Torelli R, Fiori B, Santangelo R, et al. Role of AFR1, an ABC transporter-encoding gene, in the in vivo response to fluconazole and virulence of Cryptococcus neoformans. Infect Immun 2006; 74:1352 - 9; http://dx.doi.org/10.1128/IAI.74.2.1352-1359.2006; PMID: 16428784
- Takahashi S, Kudoh A, Okawa Y, Shibata N. Significant differences in the cell-wall mannans from three Candida glabrata strains correlate with antifungal drug sensitivity. FEBS J 2012; 279:1844 - 56; http://dx.doi.org/10.1111/j.1742-4658.2012.08564.x; PMID: 22404982
- Netea MG, Ferwerda G, van der Graaf CA, Van der Meer JW, Kullberg BJ. Recognition of fungal pathogens by toll-like receptors. Curr Pharm Des 2006; 12:4195 - 201; http://dx.doi.org/10.2174/138161206778743538; PMID: 17100622
- Denning DW. Echinocandin antifungal drugs. Lancet 2003; 362:1142 - 51; http://dx.doi.org/10.1016/S0140-6736(03)14472-8; PMID: 14550704
- Wheeler RT, Fink GR. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog 2006; 2:e35; http://dx.doi.org/10.1371/journal.ppat.0020035; PMID: 16652171
- Wheeler RT, Kombe D, Agarwala SD, Fink GR. Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. PLoS Pathog 2008; 4:e1000227; http://dx.doi.org/10.1371/journal.ppat.1000227; PMID: 19057660
- Ben-Ami R, Garcia-Effron G, Lewis RE, Gamarra S, Leventakos K, Perlin DS, et al. Fitness and virulence costs of Candida albicans FKS1 hot spot mutations associated with echinocandin resistance. J Infect Dis 2011; 204:626 - 35; http://dx.doi.org/10.1093/infdis/jir351; PMID: 21791665
- Ben-Ami R, Kontoyiannis DP. Resistance to echinocandins comes at a cost: the impact of FKS1 hotspot mutations on Candida albicans fitness and virulence. Virulence 2012; 3:95 - 7; http://dx.doi.org/10.4161/viru.3.1.18886; PMID: 22286697
- Perlin DS. Current perspectives on echinocandin class drugs. Future Microbiol 2011; 6:441 - 57; http://dx.doi.org/10.2217/fmb.11.19; PMID: 21526945
- Pfaller MA, Diekema DJ, Andes D, Arendrup MC, Brown SD, Lockhart SR, et al, CLSI Subcommittee for Antifungal Testing. Clinical breakpoints for the echinocandins and Candida revisited: integration of molecular, clinical, and microbiological data to arrive at species-specific interpretive criteria. Drug Resist Updat 2011; 14:164 - 76; http://dx.doi.org/10.1016/j.drup.2011.01.004; PMID: 21353623
- Pfaller MA, Diekema DJ, Andes D, Arendrup MC, Brown SD, Lockhart SR, et al, CLSI Subcommittee for Antifungal Testing. Clinical breakpoints for the echinocandins and Candida revisited: integration of molecular, clinical, and microbiological data to arrive at species-specific interpretive criteria. Drug Resist Updat 2011; 14:164 - 76; http://dx.doi.org/10.1016/j.drup.2011.01.004; PMID: 21353623
- Wiederhold NP. Paradoxical echinocandin activity: a limited in vitro phenomenon?. Med Mycol 2009; 47:Suppl 1 S369 - 75; http://dx.doi.org/10.1080/13693780802428542; PMID: 19255904
- Walker LA, Gow NA, Munro CA. Fungal echinocandin resistance. Fungal Genet Biol 2010; 47:117 - 26; http://dx.doi.org/10.1016/j.fgb.2009.09.003; PMID: 19770064
- Walker LA, Munro CA, de Bruijn I, Lenardon MD, McKinnon A, Gow NA. Stimulation of chitin synthesis rescues Candida albicans from echinocandins. PLoS Pathog 2008; 4:e1000040; http://dx.doi.org/10.1371/journal.ppat.1000040; PMID: 18389063
- Stevens DA, Ichinomiya M, Koshi Y, Horiuchi H. Escape of Candida from caspofungin inhibition at concentrations above the MIC (paradoxical effect) accomplished by increased cell wall chitin; evidence for beta-1,6-glucan synthesis inhibition by caspofungin. Antimicrob Agents Chemother 2006; 50:3160 - 1; http://dx.doi.org/10.1128/AAC.00563-06; PMID: 16940118
- Bizerra FC, Melo AS, Katchburian E, Freymüller E, Straus AH, Takahashi HK, et al. Changes in cell wall synthesis and ultrastructure during paradoxical growth effect of caspofungin on four different Candida species. Antimicrob Agents Chemother 2011; 55:302 - 10; http://dx.doi.org/10.1128/AAC.00633-10; PMID: 21060107
- Wiederhold NP, Kontoyiannis DP, Prince RA, Lewis RE. Attenuation of the activity of caspofungin at high concentrations against candida albicans: possible role of cell wall integrity and calcineurin pathways. Antimicrob Agents Chemother 2005; 49:5146 - 8; http://dx.doi.org/10.1128/AAC.49.12.5146-5148.2005; PMID: 16304189
- Shields RK, Nguyen MH, Du C, Press E, Cheng S, Clancy CJ. Paradoxical effect of caspofungin against Candida bloodstream isolates is mediated by multiple pathways but eliminated in human serum. Antimicrob Agents Chemother 2011; 55:2641 - 7; http://dx.doi.org/10.1128/AAC.00999-10; PMID: 21422223
- Chamilos G, Lewis RE, Albert N, Kontoyiannis DP. Paradoxical effect of Echinocandins across Candida species in vitro: evidence for echinocandin-specific and candida species-related differences. Antimicrob Agents Chemother 2007; 51:2257 - 9; http://dx.doi.org/10.1128/AAC.00095-07; PMID: 17438060
- Stevens DA, White TC, Perlin DS, Selitrennikoff CP. Studies of the paradoxical effect of caspofungin at high drug concentrations. Diagn Microbiol Infect Dis 2005; 51:173 - 8; http://dx.doi.org/10.1016/j.diagmicrobio.2004.10.006; PMID: 15766602
- Stevens DA. Frequency of paradoxical effect with caspofungin in Candida albicans. Eur J Clin Microbiol Infect Dis 2009; 28:717; http://dx.doi.org/10.1007/s10096-008-0688-y; PMID: 19130103
- Bayegan S, Majoros L, Kardos G, Kemény-Beke A, Miszti C, Kovacs R, et al. In vivo studies with a Candida tropicalis isolate exhibiting paradoxical growth in vitro in the presence of high concentration of caspofungin. J Microbiol 2010; 48:170 - 3; http://dx.doi.org/10.1007/s12275-010-9221-y; PMID: 20437148
- Kurtz MB, Abruzzo G, Flattery A, Bartizal K, Marrinan JA, Li W, et al. Characterization of echinocandin-resistant mutants of Candida albicans: genetic, biochemical, and virulence studies. Infect Immun 1996; 64:3244 - 51; PMID: 8757860
- Garcia-Effron G, Kontoyiannis DP, Lewis RE, Perlin DS. Caspofungin-resistant Candida tropicalis strains causing breakthrough fungemia in patients at high risk for hematologic malignancies. Antimicrob Agents Chemother 2008; 52:4181 - 3; http://dx.doi.org/10.1128/AAC.00802-08; PMID: 18794386
- Lee KK, Maccallum DM, Jacobsen MD, Walker LA, Odds FC, Gow NA, et al. Elevated cell wall chitin in Candida albicans confers echinocandin resistance in vivo. Antimicrob Agents Chemother 2012; 56:208 - 17; http://dx.doi.org/10.1128/AAC.00683-11; PMID: 21986821
- Kartsonis N, Killar J, Mixson L, Hoe CM, Sable C, Bartizal K, et al. Caspofungin susceptibility testing of isolates from patients with esophageal candidiasis or invasive candidiasis: relationship of MIC to treatment outcome. Antimicrob Agents Chemother 2005; 49:3616 - 23; http://dx.doi.org/10.1128/AAC.49.9.3616-3623.2005; PMID: 16127030
- Wiederhold NP, Najvar LK, Bocanegra RA, Kirkpatrick WR, Patterson TF. Caspofungin dose escalation for invasive candidiasis due to resistant Candida albicans. Antimicrob Agents Chemother 2011; 55:3254 - 60; http://dx.doi.org/10.1128/AAC.01750-10; PMID: 21502632
- Angiolella L, Micocci MM, D’Alessio S, Girolamo A, Maras B, Cassone A. Identification of major glucan-associated cell wall proteins of Candida albicans and their role in fluconazole resistance. Antimicrob Agents Chemother 2002; 46:1688 - 94; http://dx.doi.org/10.1128/AAC.46.6.1688-1694.2002; PMID: 12019077
- Anderson JB. Evolution of antifungal-drug resistance: mechanisms and pathogen fitness. Nat Rev Microbiol 2005; 3:547 - 56; http://dx.doi.org/10.1038/nrmicro1179; PMID: 15953931