Abstract
The interaction between proteins is one of the most important features of protein functions. In general, the protein-protein interactions (PPIs) network of an organism is very complex, consisting of huge amount of PPIs. Functional modules can be identified from the complex protein interaction networks. It follows that the investigation of functional modules will generate a better understanding of cellular organization, processes and functions. However, it is a great challenge to apply modularity analysis to under-studied organism, even though this organism has already been sequenced, as there are few or none experimental validated PPI data for them. Therefore, by integrating several bioinformatics methods, we provide a solution for modularity analysis of any sequenced organism. By this way, new information may be found for the organism in different level, such as protein-protein interaction, pathways or cellular process. For the computation part, it takes one to two weeks. The main impact factors are computer power and size of the PPI network. It takes longer time for the manually analysis of biological meanings of the modules.
Introduction
Most cellular processes are performed by groups of physically interacting proteins. Protein-protein interactions (PPIs) are at the heart of biological activities. A complete and reliable interaction map representing the specific binary interactions within a cell would provide a significant platform for understanding many biochemically relevant processes.
Several high-throughput experimental methods, such as pull down, immuno-precipitation, two-hybrid system and protein chips, have been developed to detect the protein-protein interactions among all proteins encoded by a genome. While the data from these experimental approaches has been useful to biologists, several shortcomings exist. In particular, the results from high-throughput interaction mappings have low accuracy, and even reliable techniques can generate many false positives when applied genome-wide. Estimated error rates of high-throughput interaction results range from 41% to 90%. Experimental interaction detection is also labor intensive and costly, in part because the number of possible protein-protein interactions is very large.
Computational methods provide a complementary approach to detecting protein-protein interactions and extending protein interactomes. A variety of computational methods have been applied to observe or predict the PPI networks in biological systems. These methods enable one to discover novel putative interactions and often provide information for designing new experiments for specific protein sets. The computational approaches for in silico prediction can be classified into several general categories: genomic scale approaches, sequence-based approaches, structure-based approaches, protein domain-based approaches, learning-based approaches and network-topology-based approaches.
However, obtaining networks of PPIs is not the final target. A great challenge is how to manage and analyze the huge number of data on PPIs. It has been reported that the metabolic networks of 43 distinct organisms are organized into many small, highly connected topological modules that combine in a hierarchical manner into larger, less cohesive units.Citation1 A module of a PPI network may represent a protein complex, or a group of proteins participating in the same cellular process. The prediction and analysis of PPI modules will aid us in illustrating the basic mechanisms of biological activities
Cluster analysis is an obvious choice for the extraction of functional modules from networks of PPI. Clustering can be defined as the grouping of objects based on their shared discrete, measurable properties. A variety of clustering algorithms have been developed and successfully used in diverse fields. Recently, a systematic quantitative evaluation of the four most important clustering algorithms has been presented by Brohee and Van Helden.Citation2 The four algorithms were RNSC, MCODE, SPC and Markov Cluster algorithm (MCL). Their results showed that the MCL algorithm was both remarkably robust to graph alterations and superior for the extraction of complexes from interaction networks.
In our published paper,Citation3 we aimed to find out a way to analysis on the modularity of sequenced organism’s PPI network, especially pathogens. We also want to see what can be interpreted on pathogenicity and cellular process by the modularity analysis.
We selected EHEC O157:H7 Sakai strain in our study. EHEC O157:H7 causes diarrhea and hemolytic uremic syndrome (HUS), which is a worldwide threat to public health and has been implicated in many outbreaks of the hemorrhagic colitis. The Sakai strain of EHEC O157:H7 was sequenced in 2001 by Japan.Citation4
In our study, first, a domain-based method was used to predict the O157:H7’s PPI. Then we used the Markov Cluster algorithm (MCL) and separated 172 modules out of the O157 PPIs predicted above. After evaluation, we found that most of the modules were functional homogeneous and biologically significant. One hundred and twenty one modules were considered highly reliable, which may provide directions for experiment research. Six pathogenicity-related modules were analyzed, some of which are new and deserve further experimental validation. After investigation of relations among modules, modularity of cellular function and cooperative effect are discussed. In view of modules, our modularity analysis can give a better understanding of cell function. Moreover, predicted modules can provide possible candidates for biological pathway extension and clues for discovering new cross-talks between biological pathways. Overall, these results provide the first modularity analysis of a pathogen and shed new light on the study of pathogenicity and cellular process.
Materials
Reagents
Bioinformatics data, other than reagents, is used in the protocol, as it is a bioinformatics study. The experimentally determined interactions data of proteins are downloaded from DIP database. Sequences for the proteins of E. coli O157:H7 are downloaded from the NCBI Refseq database in Fasta format.
Equipment
Computers with Unix/Linux and Windows XP systems are required. Computer with high performance in calculation is also recommended, as there are high computation loads for this protocol.
Procedure
is a simple process map of the protocol.
Figure 1. A flowchart of the protocol.
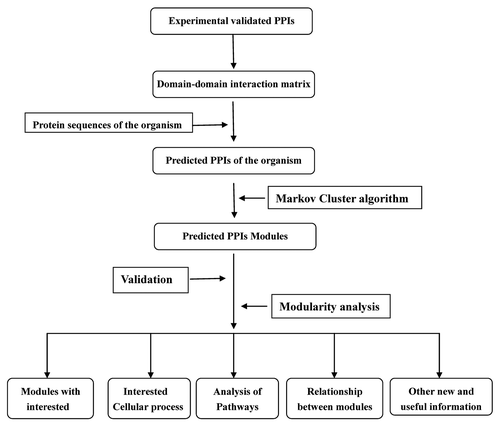
Prediction of PPIs
Maximum likelihood estimation (MLE)Citation5 and maximum specificity set cover (MSSC)Citation6 are both based on the Association Method (AM). These methods use currently available protein-protein interaction data, and estimate the probabilities of domain-domain interaction observed in PPIs. Then using the inferred domain-domain interaction, these methods can predict previously unknown protein interaction. As MLE and MSSC modify AM in a different and independent way to improve accuracy, we combined these two methods to achieve a better result. We adopted 3,722 creditable protein interactions from DIP database (data set downloaded in November 2007), which are validated by two or more experimental methods. These 3,722 PPIs were selected from 54,511 PPIs deposited in DIP database referring to more than 200 organisms, including both non-pathogens and pathogens.
A domain interaction matrix is built using the MLE-MSSC method based on the credible proteins above. Then, all proteins of the selected organism are scanned by InterProScan program to obtain the domains of these proteins. The total matched pairs of the selected organism’s proteins were compared with the domain interaction matrix, and raw predicted PPIs data were obtained. Two post-processing steps were applied to the raw PPI data. First, we eliminated directional repeats from the PPIs. Because the prediction program cannot predict weighted directional PPIs, directional PPIs are actually the same. Second, we eliminated self-interactions. The existence of self-interactions will generate single protein modules when using the MCL algorithm to predict protein modules. Although simple protein modules may represent homogeneous multimers, without additional information, these would be difficult to analyze. shows the predicted data of O157 protein interactions.
Table 1. Predicted data of O157 protein interactions
Prediction of modules from PPIs network
Markov Cluster algorithm (MCL) is used to predict modules from the PPIs obtained above, with an Inflation coefficient of I = 1.8. However, the modules predicted by the MCL algorithm have no overlapping components, while in real organisms, there are some proteins that exist in multiple complexes, or participate in several cellular processes at the same time. So, we identified the proteins shared between modules by a post-processing step. In this step, proteins assigned to each cluster (donor cluster) were scanned for interaction partners in other clusters, and proteins interacting with a sufficiently large fraction of partners in another cluster (acceptor cluster) were also assigned to that cluster. Comparison with known or conserved protein complex, KEGG pathways and Go annotation analysis are recommended for evaluation of the quality of the predicted module.
Analysis of predicted modules
If you have any interested proteins, for example proteins related to pathogenicity, you can locate and find the modules containing these proteins. The interactions inside the module can be displayed by visualization tools such as Pajek or Cytoscape. Therefore, more information about the interested protein or the biological process can be found by analysis the interaction map. Also it is easy to investigate the relationship between modules with similar biological function.
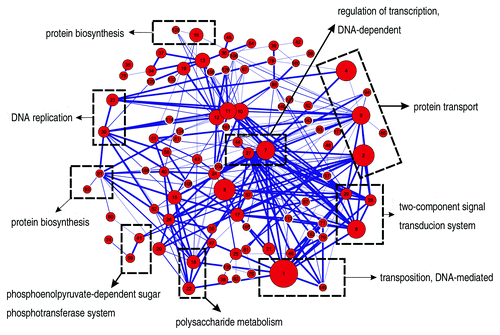
In our study, 172 modules are obtained from the O157 PPIs predicted above. After evaluation, we found that most of the modules were functional homogeneous, and biologically significant. One hundred and twenty-one modules were considered highly reliable, which may provide directions for experiment research. Six pathogenicity-related modules were analyzed, some of which are new and deserve further experimental validation. After investigation of relations among modules (), modularity of cellular function and cooperative effect are discussed. In view of modules, our modularity analysis can give a better understanding of cell function. Moreover, predicted modules can provide possible candidates for biological pathway extension and clues for discovering new cross-talks between biological pathways.
Figure 2. Relations among predicted modules. Red node in the network represents a module. Node radius is proportional to the module’s size. Node labels represent module number. Edges represent at least two PPIs among modules. Modules in dashed rectangle have same GO biological process term with each other.
Timing
For the computation part, prediction of PPIs and protein modules takes one to two weeks. The main impact factors are computer power and size of the PPIs network. It takes a longer time for the manual analysis of biological meanings of the modules.
Problem Handling
Quality evaluation of predicted PPIs
The reliability of the predicted PPIs is the fundament of this research, as the module prediction and further analysis all base on these data. Although MLE and MSSC have been proved of good sensitivity and specificity in their original papers, we still recommend several methods below to validate the reliability.
The best way to evaluate the predicted PPIs is to compare and see the overlap with the experimental validated the PPIs. However, for under-studied organisms, there are not enough experimental validated PPIs available. So we choose to compare the PPIs data in the STRING database, which collects experimental and predicted protein-protein interactions. The PPIs in STRING database include direct and indirect (functional) associations, which are derived from four sources: genomic context, high-throughput experiments, coexpression and previous knowledge. The percent of predicted PPIs that overlap with the PPIs in STRING database can be used as one criterion here to evaluate the quality. In general, overlap ~20% will be good enough, as protein-protein interactions are dynamic, and result of different assays or even same assays in different time or condition varies. Even the overlap of high-throughput experimental yeast interactome by GavinCitation7 and KroganCitation8 is ~25%.
Topological analysis of predicted PPIs network is also useful to evaluate the quality. Yook et al. compared four available databases that represent the protein interaction network of S. cerevisiae and found that the yeast protein interaction network in each database is of scale-free topology and hierarchical modularity.Citation9 Li et al. analyzed three of the largest protein interaction network of S. cerevisiae, C. elegans and D. melanogaster and also confirmed the scale-free, small-world property.Citation10 So we can analyze and see if the predicted PPIs network have the same characterization with those networks obtained by experiments.
GO distance and semantic similarity are also good options to evaluate the quality. For GO distance, the smaller the value, the more specific category the two proteins belong to and thus they are more likely to interact. While for GO semantic similarity, the higher the value, the more likely the proteins to interact. We can compute these values of the predicted PPIs data set, and compare with the random obtained data set to see if it is statistical significant.
Calculation in the protocol
Programming background is required for this protocol. As in general, there are many data to handle in the study. It is better to use script (like Perl) to automatically do the computation, data mapping and statistical analysis, etc.
Anticipated Results
This protocol provides necessary information to modularity analysis of a sequence organism. First, PPIs can be predicted by a domain-based method. Then modules can be separated out of the predicted PPIs. Further analysis of the predicted modules may provide directions for experiment research, and give a better understanding of cell function and biological pathways.
References
- Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabási AL. Hierarchical organization of modularity in metabolic networks. Science 2002; 297:1551 - 5; http://dx.doi.org/10.1126/science.1073374; PMID: 12202830
- Brohée S, van Helden J. Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics 2006; 7:488; http://dx.doi.org/10.1186/1471-2105-7-488; PMID: 17087821
- Wang X, Yue J, Ren X, Wang Y, Tan M, Li B, et al. Modularity analysis based on predicted protein-protein interactions provides new insights into pathogenicity and cellular process of Escherichia coli O157:H7. Theor Biol Med Model 2011; 8:47; http://dx.doi.org/10.1186/1742-4682-8-47; PMID: 22188601
- Hayashi T, Makino K, Ohnishi M, Kurokawa K, Ishii K, Yokoyama K, et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res 2001; 8:11 - 22; http://dx.doi.org/10.1093/dnares/8.1.11; PMID: 11258796
- Deng M, Mehta S, Sun F, Chen T. Inferring domain-domain interactions from protein-protein interactions. Genome Res 2002; 12:1540 - 8; http://dx.doi.org/10.1101/gr.153002; PMID: 12368246
- Huang CB, Morcos F, Kanaan SP, Wuchty S, Chen DZ, Izaguirre JA. Predicting protein-protein interactions from protein domains using a set cover approach. IEEE/ACM Trans Comput Biol Bioinform 2007; 4:78 - 87; http://dx.doi.org/10.1109/TCBB.2007.1001; PMID: 17277415
- Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006; 440:631 - 6; http://dx.doi.org/10.1038/nature04532; PMID: 16429126
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 2006; 440:637 - 43; http://dx.doi.org/10.1038/nature04670; PMID: 16554755
- Yook SH, Oltvai ZN, Barabási AL. Functional and topological characterization of protein interaction networks. Proteomics 2004; 4:928 - 42; http://dx.doi.org/10.1002/pmic.200300636; PMID: 15048975
- Li D, Li J, Ouyang S, Wang J, Wu S, Wan P, et al. Protein interaction networks of Saccharomyces cerevisiae, Caenorhabditis elegans and Drosophila melanogaster: large-scale organization and robustness. Proteomics 2006; 6:456 - 61; http://dx.doi.org/10.1002/pmic.200500228; PMID: 16317777